 Partha S. Saha*
Partha S. Saha* William G. Mayhan
William G. Mayhan- Division of Basic Biomedical Sciences, Sanford School of Medicine, University of South Dakota, Vermillion, SD, United States
Alcohol is a well-known teratogen, and prenatal alcohol exposure (PAE) leads to a greater incidence of many cardiovascular-related pathologies. Alcohol negatively impacts vasculogenesis and angiogenesis in the developing fetal brain, resulting in fetal alcohol spectrum disorders (FASD). Ample preclinical evidence indicates that the normal reactivity of cerebral resistance arterioles, which regulate blood flow distribution in response to metabolic demand (neurovascular coupling), is impaired by PAE. This impairment of dilation of cerebral arteries may carry implications for the susceptibility of the brain to cerebral ischemic damage well into adulthood. The focus of this review is to consolidate findings from studies examining the influence of PAE on vascular development, give insights into relevant pathological mechanisms at the vascular level, evaluate the risks of ethanol-driven alterations of cerebrovascular reactivity, and revisit different preventive interventions that may have promise in reversing vascular changes in preclinical FASD models.
Introduction
One of the most often used psychotropic substances, ethanol, permeates the placental barrier, and hence exerts potential teratogenic effects. A 2011 survey found that 45 percent of pregnancies in the United States were unintentional, with one in ten pregnant women between the ages of 18 and 44 reporting intake of alcohol in the previous 30 days and one in thirty-three reporting binge drinking [1, 2]. The pattern and intensity of the detrimental effects of prenatal alcohol exposure are dependent on alcohol dose, timing, sequence, and persistence of alcohol intake. All current research indicates that alcohol has a detrimental effect on fetal development. However, the fetal brain is the most substantially afflicted organ, displaying structural and functional abnormalities as a result of maternal alcohol consumption [3, 4]. Exposure to alcohol may result in fetal alcohol spectrum disorders (FASDs) in humans. FASD is a collective name for the harmful effects of prenatal alcohol exposure (PAE), including impairments in physiologic, neurologic, and behavioral development [5]. It should also be noted that FASD is an umbrella term that incorporates a variety of alcohol-related disorders with a range of severity. The most severe type of FASD, fetal alcohol syndrome (FAS) is characterized by specific physical traits in neonates. Partial FAS (pFAS), on the other hand, is diagnosed when there is validated prenatal alcohol exposure but not enough signs and symptoms to confirm a diagnosis of FAS [6]. Recent investigations, however, have seen the effects of alcohol on fetal cerebrovascular function emerge as a key mediator since brain energy needs are usually fulfilled by a continually adjusting blood supply [7]. Such an adaptation begins at the level of cerebral arteries and extends to microvessels that enter the brain parenchyma and form the neurovascular unit [8,9]. PAE may have acute and chronic teratogenic and toxic impacts on cerebrovascular physiology. This review’s objective is to outline the pathological pathways in different developmental phases, as well as their morphological and functional consequences on the cerebral circulation. It is held that alterations of cerebral blood flow (CBF) owing to dysregulation of cerebral blood vessels in PAE may be a significant contributor to the etiology of several cerebrovascular events, such as stroke.
Cerebrovascular effects of PAE during fetal development
During the early-gestation period, the neural tube is surrounded by a perineural vascular plexus (PNPV) developed by vasculogenesis, the process of blood vessel development in the embryo, which involves the de novo generation of endothelial cells (ECs). Beginning in the mid-gestation period, endothelial cells from the PNPV penetrate the neural parenchyma, utilizing fibers from multipotent stem cells, like radial glia cells from the developing forebrain, to direct their migration toward the ventricular surface, thereby initiating the first vessels of the CNS. The second trimester is crucial for neuronal and blood vessel growth in the fetal brain [10, 11]. During this phase, a network of arteries inside the sub-arachnoid space gives birth to microvessels that enter the fetal brain [11]. This emerging vasculature supports nutritional requirements and endocrine regulation of fetal brain development [12]. In the early-mid gestation period, vascular development in the CNS is mediated by angiogenesis, which is defined as a series of cellular and molecular processes including the production of new vessels and culminates in the formation of the blood-brain barrier (BBB) [13,14,15]. At the mid-late gestation period, interactions among neural cells and ECs are initiated and continue through late-gestation and until birth. Communication between glial cells and the vasculature is crucial for the optimal development of the nervous system [16]. Pericytes increase vascular stability by releasing a variety of stabilization factors, such as angiopoietin-1 (ANG1) and platelet-derived growth factor (PDGF-β), tissue inhibitor of metalloproteinase 3 (TIMP3) [17]. In response to neural impulses at the perivascular terminal, astrocytes produce chemicals capable of regulating vascular tone. From the late-gestation period until birth, the BBB continues to mature via the expression of various molecules by the BBB’s cellular components, culminating in the development of a basal membrane rich in laminin, collagen IV, and fibronectin that fully surrounds the brain vasculature [18].
It has been established that maternal ethanol exposure causes rapid and persistent loss of blood flow from the umbilical artery to the fetal brain, potentially distressing nutrition and the maternal/fetal endocrine environment during a critical stage for neurogenesis and angiogenesis in the developing brain [19]. Because alcohol exposure during pregnancy is known to alter brain development, the majority of investigations have centered on neural cells. At the molecular and subcellular levels, however, cerebrovascular development is a more complicated, multi-step process involving a large number of molecular participants.
Angiogenesis is a complex process that is delicately controlled by a balance between pro-and anti-angiogenic factors [20]. They consist of, but are not limited to, vascular endothelial growth factor (VEGF), integrins, fibronectin, angiopoietins, vascular cell adhesion molecules (VCAM), fibroblast growth factors (FGF), tyrosine kinases (TK), extracellular matrix (ECM) proteins and proteinases, transforming growth factor (TGF), and Wnt signaling factors as growth-stimulatory signals. On the other hand, tumor necrosis factor (TNF) signaling and pro-apoptosis factors are stop signals [21, 22, 23]. Recent research conducted on adult nonhuman primates [24] and mice [25, 26] conclusively demonstrates that alcohol has a deleterious effect on vasculogenesis and angiogenesis pathways. According to microarray investigations, genes (e.g., VEGF gene family) implicated in angiogenesis are molecular targets of ethanol toxicity [27].
In addition to disrupting the signaling components that regulate vascularization and brain development, PAE may affect molecular targets that are particular to cerebrovascular development. Ethanol may affect the molecular and cellular elements of the BBB directly as soon as they are present during CNS development. Other research indicated that the embryonic brain is more susceptible to ethanol due to the high prevalence of proapoptotic proteins and low expression of proteins associated with stress response systems, namely autophagy or unfolded protein response [28]. Also, a proteomics investigation revealed that alcohol-exposed baboon fetuses mainly had changes in mitochondrial and structural proteins in their cerebral arteries. Unlike mitochondrial proteins, structural proteins were downregulated in the brain of fetal arteries exposed to alcohol [29].
Morphological damage in cerebral blood vessels due to PAE
Considering ethical limits of human-based research, the PAE of laboratory animals has been extensively used as an alternative. Results from these animal models showed a significant influence on the generation and expression of microvessels in the rodent model. Besides dose, the timing of ethanol exposure has a significant impact on the outcome of fetal brain development. Since the gestational period of rodents (i.e., 18–23 days) is much shorter than that of humans, the morphological and functional effects of alcohol on these animals may need to be extrapolated to equivalent pregnancy terms in humans. In both rats and mice, gestational day (GD) 1–10, GD 10–20, and postnatal days (P) 1–10 can be considered as the equivalents of first, second, and third trimester human pregnancy, respectively [30]. Prior research on rat models exposed to moderate prenatal alcohol dosages revealed ultrastructural changes in brain capillaries at P20–30 [31]. In a study using rat models, the oral administration of 6.6 g/kg of ethanol from P4 to P10 was evaluated at P10 for any alterations in brain microvasculature [32]. There was no change in capillary density, while capillary diameters were increased in the cerebellum and hippocampus regions, unlike the dentate gyrus region [32]. Later, in a mouse model (at P2) of the third trimester equivalent of human pregnancy demonstrated the loss of radial orientation of the microvessels and a reduction in cerebral vascular density when maternal injection of 3 g/kg ethanol occurred during GD 13–19, which was a time-line equal to the second trimester of human pregnancy [33]. Overall, these rodent studies suggest that alcohol exposure during the human mid and late trimesters might affect the microvasculature differentially in the brain regions.
Human embryos are susceptible to ethanol. Evidence suggests that exposure to high amounts of ethanol during human development causes craniofacial, cardiovascular, and neurological abnormalities, which are often accompanied by cognitive and behavioral deficiencies. Autopsy and magnetic resonance imaging (MRI) investigations found that individuals who were exposed to alcohol in utero had structural brain damage, including lower brain sizes and reduced amounts of white and gray matter inside the brain [reviewed in 4]. However, the animal research mentioned earlier suggests that exposure to PAE during late gestation might be perilous. In a postmortem examination of human brain tissues from fetuses that deceased spontaneously in utero, stage-dependent alterations in the cortical vascular network were detected in the cortex of fetuses with FAS. The radial arrangement of cortical microvessels was markedly disrupted in FAS patients after 30 weeks of gestation, whereas no changes were seen in alcohol-exposed human embryonic brain tissues between 20 and 22 weeks of gestation. In addition, dynamic microscopy techniques indicated that alcohol altered endothelial cell activity and survival as well as the plasticity of the microvessel [33].
In conclusion, investigations conducted on rodents and humans suggest that prenatal alcohol consumption, especially during the late stages of gestation, may affect the macroscopical or microscopical structure of the cerebral microvascular network. These age-dependent abnormalities can be theorized because of a disturbance in cortical angiogenesis.
Functional changes in cerebrovascular circulation due to PAE and their possible implications
Critical to brain function is a coherence among the metabolic needs, the supply of oxygen and nutrients, and the elimination of cellular waste. This matching requires continual modulation of CBF, which may be divided into four basic categories: autoregulation, vascular reactivity, neurovascular coupling (NVC), and endothelium-dependent responses. Due to the limited scope of this paper, this part of the review will discuss how PAE affects the functional parts of cerebrovascular circulation, with focuses on how it changes CBF and vascular reactivity and the clinical effects of these changes.
Changes in CBF
The effects of maternal alcohol intake during pregnancy on outcome depends on the quantity and patterns of alcohol consumption. In animal trials, binge-like drinking patterns, in which the unborn is exposed to elevated blood alcohol concentrations (BACs) during relatively brief intervals, were shown to be more detrimental, even when the overall quantity of alcohol taken was lower than that of more continual drinking patterns [34, 35, 36]. Because binge drinking produces high BACs, may occur at important phases of brain development, and can be coupled with recurrent withdrawal episodes, it may be extremely detrimental [35]. In addition to a single binge pattern, recurrent binge-like events of maternal ethanol consumption are harmful to the fetus likewise [35] and may cause transitory and chronic abnormalities in cerebrovascular functioning. Abrupt CBF alterations in embryos may be associated with craniofacial deformity, fetal growth limitation, neuronal death, impaired delivery of nutrients to and elimination of metabolites from neurons, and a reduction in cerebral arterial tone. Here, we will examine how varied patterns of maternal alcohol use affect the blood flow to the cerebral arteries.
Temporary effects of maternal alcohol consumption on CBF
Following acute ethanol consumption, previous research on pregnant murine [19], ovine [41, 40, 39, 38, 37], and baboon [46, 45, 44, 43, 42] have showed aberrant uterine and cerebral blood flow. During acute maternal alcohol intake in vivo, the majority of investigations observed an increase in fetal cerebral perfusion and, perhaps, a decrease in fetal cerebral artery blood flow doppler velocity indices [46, 38, 42, 19]. In contrast, maternal infusions of 1 g/kg ethanol were associated with a reduction in CBF in preterm sheep [39] and baboons [45]. There was no decline in fetal CBF in mid-gestation sheep [40]. In this trial, 1 g/kg of ethanol was infused into the maternal blood at gestational day 92 (human equivalent term at 145–150 days), resulting in a maternal BAC of 150 mg/dl [40]. In contrast, in a separate experiment using the same animal model and a comparable experimental technique, Parnell et al. (2007) found that a greater dosage of ethanol (1.75 g/kg) substantially enhanced CBF by over 30 percent. Specifically, in the cerebellum, the rise in CBF 1 h after ethanol infusion was up to 50 percent greater than in the control group. These two experiments together demonstrated that, in vivo, the effects of alcohol on cerebral blood flow were concentration-dependent and brain region-specific.
In addition, the alterations in fetal CBF were detected along with the changes in systemic hemodynamics: a considerable increase in fetal cardiac output and heart rate, as well as a decrease in mean arterial pressure and systemic peripheral resistance [41]. An ultrasonography investigation demonstrates that acute single and recurrent binge-like episodes of maternal ethanol intake may promptly and chronically alter cranially-directed fetal blood flow throughout the second trimester [19]. PAE not only induces alterations in CBF but also impairs autoregulation. Additionally, PAE also modulates CBF responses to environmental variables. It was shown that fetal cerebral vasodilator responses to hypoxia [47] and acidosis [48] are altered by exposure to ethanol during the second trimester. Earlier work in an ovine model of pregnancy showed that PAE (1 g/kg ethanol maternal infusion i.v. for 3 weeks during the equivalent of the first trimester) reduced the adaptive rise in CBF in response to hypoxia in 1–4-day-old term lambs. As a consequence, neonatal brain oxygen supply could not be sustained [49]. Thus, it demonstrates that PAE as early as the first trimester might increase cerebral vascular susceptibility to environmental damage. Changes in the blood’s metabolic profile, such as acidemia and hypercapnia, often follow acute alcohol-induced disruption of CBF and may contribute to its clinical effects [41, 40, 39].
Persistent effect of maternal alcohol consumption on CBF: Impairment of vascular reactivity
Multiple groups have explored the influence of PAE on fetal cerebrovascular function. A study, utilizing doppler ultrasonography revealed that in a baboon model of pregnancy, diminution of PAE alcohol impact occurred earlier to delivery. That study particularly indicated that acute fetal alcohol intake, with a maternal BAC of 80 mg/dl in the equivalent of the second trimester of human pregnancy, decreased cerebral blood flow in fetal cerebral arteries. However, this influence on fetal vascular function did not persist throughout the duration of pregnancy [45]. This indicates that alcohol-induced changes in the physiological characteristics of the fetal cerebral arteries disappear with development. In another example of an ovine PAE model, the alcohol impact of PAE diminished after delivery. Adult cerebral artery dilatory effects to adenosine A2A receptor agonist, CGS21680, were investigated in vitro using arterioles extracted from third trimester comparable ovine fetuses exposed to ethanol in utero. The dilatory reaction to micromolar doses of CGS21680 was substantially greater when compared to control group [48]. However, when arterioles were collected from adult sheep using a similar ethanol administration approach [50], same dilator responses were comparable to the control counterpart. Therefore, experiments using diverse animal models suggested that PAE-induced abnormalities in the pharmacological nature of cerebral arteries were slowly reversed with aging.
Turcotte et al. found that prenatal exposure to ethanol (6.4%) lowered the relaxation of the aorta to carbamylcholine in rats 25 weeks after birth, indicating a change in the reactivity of peripheral arteries [51]. Similarly, in peripheral arteries, some of the alcohol-induced modifications in fetal cerebral artery characteristics may persist and be detectable long after birth. Multiple studies on rats reveal the long-lasting effects of PAE. In vivo responses of cerebral arterioles to eNOS- and nNOS-dependent agonists were reduced in young rats (4–6 weeks old) [52] and in adult rats (12–15 weeks old) [53,54] exposed to alcohol during fetal development. While it is known that prenatal alcohol exposure impairs the dilatation of cerebral arterioles in rats, no research has explored the effect of prenatal alcohol on the constrictor response of cerebral arterioles until recently. The response of cerebral arterioles to U-46619 (a thromboxane-mimetic analog) and arginine vasopressin (AVP), which are physiologically important constrictors, was comparable in male and female rats independent of prenatal alcohol exposure and age. Similarly, there was no difference between male and female adolescent rats’ responses to angiotensin II after prenatal alcohol consumption. At adulthood, however, alcohol-exposed females demonstrated an unanticipated dilatation in response to a high concentration of angiotensin II, but males did not. Except in adult female rats, the majority of the vasoconstriction responses to prolonged prenatal alcohol exposure were retained [55]. Besides inducing the loss of vascular reactivity, PAE is capable of inducing arterial stiffness to the cerebral vasculatures [56].
Implications
Long-term research on humans have previously proven that offspring of binge-drinking mothers demonstrate particularly significant cognitive and behavioral abnormalities. The ability of the brain to maintain adequate cerebral blood flow in the face of changes in metabolic demand may influence the pathogenesis of symptoms associated with FASD in adults, i.e., cognitive decline, psychiatric symptoms, dementia, and seizures, all of which may be directly impacted by the ability of the brain to maintain adequate cerebral blood flow (neurovascular coupling) [57–60].
In addition, the loss of cerebrovascular autoregulation, such as during severe hypertensive encephalopathy, may result in catastrophic occurrences, such as subarachnoid hemorrhage and hemorrhagic stroke [61]. Evidently, our lab has also shown that in prenatal exposure to alcohol exacerbated brain damage in adult rats after ischemia/reperfusion and that treatment of dams with apocynin reduced this increase in brain injury following ischemia/reperfusion [53]. Thus, it may be inferred that PAE-induced alterations in cerebral blood flow not only contribute to the pathophysiology of fetal alcohol syndrome, but also have the potential to cause serious brain injury.
Molecular and cellular mechanisms of impairment and recent preclinical interventions
Because alcohol is a simple ligand that may concurrently target several chemical entities, its effects on the developing brain are very complicated. Alcohol may cause cell death in some types of brain cells while interfering with the cellular and molecular activities of other types. These effects may be caused by alcohol both directly and indirectly.
Alcohol may have direct effects on embryonic brain development by interfering with neuronal proliferation and migration [62] or by inducing cell death [63,64,65]. In addition, alcohol may raise fetal glutamate levels [66, 67] and decrease glutamate N-methyl-D-aspartate receptors [68, 69], which may result in aberrant neuronal and glial migration.
Alcohol-induced hypoxia in the fetus is a significant indirect cause of alcohol. Alcohol reduces blood flow to the umbilical artery [70, 71], which might result in growth retardation [72, 73]. In addition to inhibiting protein synthesis and altering hormone levels, alcohol may further impede development [74, 75]. Increased oxidative stress on the embryo [76, 77] and disruption of growth factor signaling [78, 79] are additional pathways.
These effects can be toxic (short-term) and teratogenic (long-term). This section will examine as well as summarize (Table 1) the toxic and teratogenic processes that generate short-term or long-term effects on the cerebral arteries and microvessel network.

TABLE 1. Multiple cellular and molecular mechanisms of cerebrovascular impairment due to prenatal alcohol exposure.
Mechanism of transient and persistent effects of prenatal alcohol exposure
Alcohol’s short-term effects on CBF are highlighted by ethanol’s targeting of molecular players inside the fetal cerebral blood vessel. In particular, PAE is characterized by fetal cerebral artery dilatation in the presence of alcohol, which is mediated by cannabinoid (CB) receptors 1 and 2. A study showed that the endocannabinoid (eCB) system is a target of maternal alcohol intake inside the fetal cerebral arteries, and that rimonabant, a CB receptor inverse agonist, might be a potential rescue treatment, as suggested by Seleverstov et al. [46]. Furthermore, fetal CB2 receptor-mediated cerebral artery dilation by anandamide was up-regulated in alcohol-exposed fetuses, showing that these receptors are the primary determinants of the persistent vasodilatory impact of alcohol on fetal cerebral arteries [46]. In addition to the increase of anandamide (AEA)-induced dilatation, other mechanisms of PAE’s lasting effects are described in subsections below.
Endothelial nitric oxide synthase
Endothelial nitric oxide synthase (eNOS) is extremely susceptible to prenatal ethanol exposure in fetal cerebral arteries. In fetal cerebral arteries of an ovine model of pregnancy - where fetal plasma alcohol concentration reached 108 mg/dl during the late gestational period of day 95–133 (human equivalent term pregnancy) - a decreased endothelium-dependent vasodilation was observed in response to the dilator 5-hydroxytriptamine in alcohol-exposed donors. Authors also discovered a substantial reduction in endothelial nitric oxide synthase (eNOS) mRNA [56]. In contrast, in adolescent rats (4–6 weeks old) and adult rats (12–15 weeks old), in utero alcohol exposure decreased cerebral arteriole responses to eNOS (ADP) and neuronal nitric oxide synthase (nNOS)-dependent (NMDA) agonists [52, 53]. However, in microvessels and tissue from the parietal cortex of adolescent rats, the expression of eNOS and nNOS levels was not altered [52].
Stiffness of the arterioles
In the ovine model mentioned above, Parkington et al. also discovered that PAE significantly increased the fetal cerebral artery’s total functional stiffness. Evidently, the elastic modulus of arteries in alcohol-exposed groups was approximately double that of control groups. In comparison to the control group, the alcohol-exposed group showed considerably higher mRNA levels for collagen Ia1 and tropoelastin, revealing the mechanism behind the enhanced functional stiffness of the vessel [56].
Increasing production of Reactive Oxygen Species (ROS)
EtOH and its catabolite acetaldehyde are itself harmful, although oxidative stress is the predominant damaging mechanism, according to current understanding. Oxidative stress is characterized by high intracellular levels of reactive oxygen species (ROS) that cause lipid, protein, and DNA damage. ROS are molecules or ions generated by the incomplete reduction of oxygen by one electron. The principal ROS species are superoxide (O2•), hydrogen peroxide (H2O2), and the hydroxyl radical (•OH). Cananzi and Mayhan evaluated the production of superoxide in the cerebral arterioles of prenatally alcohol-exposed adolescent and adult rats [52, 53], given that oxidative stress has been found to enhance neurovascular and neuronal damage and death in multiple brain locations in FASD (Figure 1A). PAE increased the baseline levels of superoxide in the parietal cortex tissue of rats (Figure 1B), even when NADPH, which was used to enhance NADPH oxidase activity, was administered. NOX-2 and NOX-4 are isoforms of the NADPH oxidase system that are found in endothelium and vascular smooth muscle and play a crucial role in the generation of superoxide [80–84]. Alcohol exposure during gestation increases the expression of NOX-2 and NOX-4 in microvessels. In addition, they discovered that apocynin, a powerful antioxidant, lowered superoxide levels (Figure 1C) and relieved impairment of eNOS- and nNOS-dependent responsiveness of cerebral arterioles in rats exposed to alcohol during gestation. This indicated an increase in superoxide generation in these animals, which may lead to an increase in oxidative stress in rats exposed to alcohol during gestation [52, 53]. Importantly, they discovered that apocynin reduces the likelihood of brain injury in adult rats after cerebral ischemia [53]. As apocynin was a nonspecific inhibitor of superoxide anion, the particular subcellular route underlying the impairment of cerebral vascular function remained unclear. Saha et al. used rosiglitazone, an agonist for the gamma subtype of peroxisome proliferator-activated receptors (PPARs) to determine the specific vascular anti-oxidant mechanism [54]. It has been shown that these receptors sit on vascular smooth muscle and endothelium [85,86,87]. In addition, both acute and chronic treatment of male and female adult rats with rosiglitazone protected decreased eNOS and nNOS-dependent vascular function in alcohol-exposed male and female adult rats. In addition, acute rosiglitazone decreased superoxide levels (Figure 1C) in parietal cortex tissue, indicating the anti-oxidant mechanism(s) via which rosiglitazone enhanced vascular function [54]. PAE (three percent ethanol for entire gestation period) induced dysfunction in the ability of specific potassium channels to dilate in rat cerebral arterioles. This dysfunction appears to be mediated by an increase in oxidative stress, as acute apocynin was able to enhance the response [unpublished data].
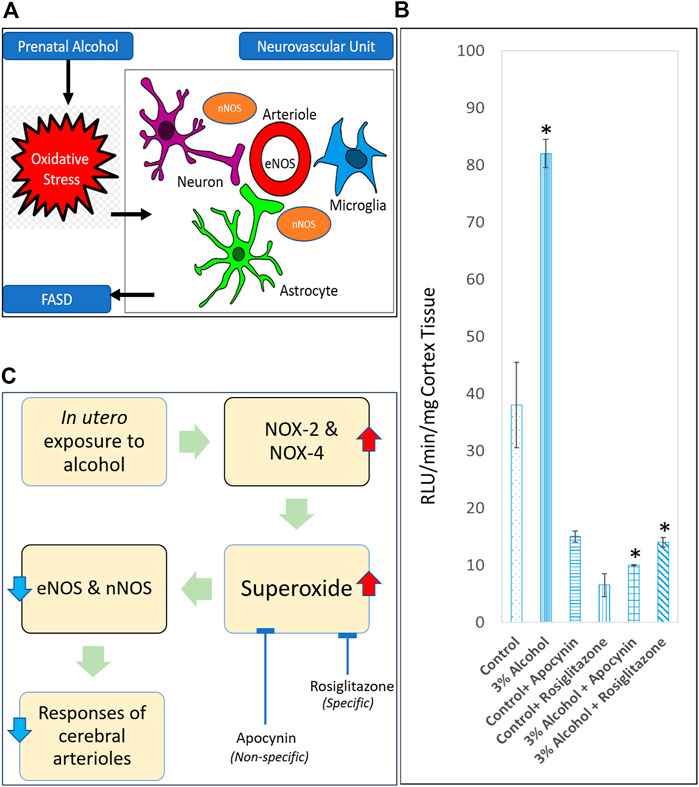
FIGURE 1. Role of oxidative stress in causing neurovascular damage in FASD. (A) Endothelial nitric oxide synthase (eNOS) and neuronal nitric oxide synthase (nNOS) are some of the targets for prenatal alcohol-induced oxidative stress in the neurovascular unit. (B) Superoxide levels at baseline in parietal cortex tissue from adult control rats, rats exposed to alcohol in utero, control + apocynin rats, control + rosiglitazone rats, alcohol + apocynin, and alcohol + rosiglitazone rats [Adapted and combined from 53 & 54 with proper permission from publisher, John Wiley and Sons]. *p < 0.05 versus levels before treatment with apocynin and rosiglitazone. (C) Proposed pathway for a physiological relationship between in utero alcohol exposure and impaired cerebral arterioles due to superoxide production. Expansion for NOX = NADPH oxidase.
Neurohormonal alteration
Vasoactive intestinal peptide (VIP) operates as a nonadrenergic, noncholinergic neurotransmitter or neuromodulator in both the peripheral and central nervous systems, where it acts on specific receptors to dilate cerebral arteries, pial arterioles, and intracerebral arterioles [88–91]. Alcohol exposure during gestation decreases VIP levels permanently in the rat fetal brain [92]. Alcohol exposure during pregnancy dramatically affected the dilator response of adult intracerebral arterioles to VIP in an adult sheep model of binge-drinking during pregnancy [50]. This decrease may suggest a loss of neuronal connections, including neurons carrying VIP, or a change in VIP receptor density in the adult brain. This is the first research to indicate that exposure to alcohol during fetal development may have long-lasting consequences on vasomotor responses in adult brain arteries.
Mechanism of modifications of morphology and integrity of NVC in response to prenatal alcohol exposure
PAE has profound impacts on angiogenesis and endothelial cell survival on the formation of cortical blood vessels. The molecular processes behind these effects are poorly understood. VEGF is an effective blood vessel development regulator. VEGF-R1, VEGF-R2, and VEGF-R3 receptors [93] mediate the biological actions of VEGFs. Jégou et al. [33] evaluated VEGF-R1 and VEGF-R2 protein levels in cerebral microvessel extracts of P2 mice exposed to prenatal alcohol in 2012. They observed an upregulation and also a simultaneous downregulation of VEGF1 and VEGF2 receptor proteins respectively in the cortical microvascular network during PAE; these changes were found to be associated with detrimental alterations in density and radial organization. Thus, disruption of these receptor subtypes is one of the mechanisms behind the VEGF-mediated modification of the cerebral microvascular network in rats prenatally exposed to alcohol. One of their in vitro experiments revealed that exogenous VEGF reduced the deleterious effect of ethanol on the vascular plasticity of the cortical glia [94].
During fetal development, well-controlled autophagy promotes vascular development and also protects against autophagic cell death. It is especially vital in ECs, one of the principal components of the developing blood vessels, as they help to adjust their bioenergetic and biosynthetic requirements in response to shifting environmental conditions, the presence of angiogenic stimuli, or intrinsic and extrinsic damages. PAE may inhibit autophagy in brain endothelial cells, hence leading to changes in angiogenesis and the resultant brain abnormalities identified in individuals with pFAS/FAS. PAE increases the frequency of autophagic vacuoles in the endothelium of cortical microvessels in human fetal brain tissues and in a mouse model of PAE in neonates, indicating defective autophagy [94, 95]. Girault et al. discovered that the levels of autophagy marker proteins, such as microtubule-associated protein 1A/1B-light chain 3 (LC3) LC3 and ubiquitin-binding protein p62, were considerably enhanced in endothelial cells treated with 50 mM ethanol in order to understand the process. In addition, a reduction in Rab7, a protein that plays a crucial function in endocytosis, was detected, which may account for the impaired autophagosome–lysosome fusion. Importantly, these effects of ethanol were eliminated in the presence of 4-methylpyrazole, which inhibits the synthesis of the ethanol metabolite acetaldehyde. These findings showed that acetaldehyde (MeCHO) triggers the process of autophagy dysregulation in cerebral microvessels after alcohol exposure [94].
In addition, it was shown that the increase in autophagy vacuoles after alcohol exposure was associated with a downregulation of the mammalian target of rapamycin (mTOR) pathway. They also demonstrated that activating autophagy with the mTOR inhibitor rapamycin reduces ethanol-induced endothelial cell death and restores vascular plasticity [94]. Overall, this shows that ethanol adversely affects angiogenesis by promoting endothelial autophagy in the cortical layer.
Previous research has shown that the neuroprotection afforded by autophagy may originate from the elimination of damaged mitochondria. And the lowered degree of autophagy may promote ROS production and excessive mitophagy [96], hence enhancing ethanol-induced cell death. In murine pulmonary microvascular endothelial cells (MPMVEC), Girault et al. found that ethanol impairs mitochondrial integrity and triggers apoptosis-inducing factor (AIF) protein release and nuclear translocation, which may result in programmed cell death. Therefore, they hypothesized that activation of autophagy by rapamycin may similarly shield endothelial cells from ethanol-induced mortality and aid cortical angiogenesis in individuals with pFAS/FAS [94].
Among numerous pathways of cell death generated by fetal brain alcohol exposure, fetal cerebral artery mitochondria-linked cellular apoptosis has been documented in an animal model of prenatal alcohol exposure and FASD-related brain injury [97]. Alcohol-induced abnormalities in prenatal cerebrovascular mitochondria may result from both direct targeting by alcohol [29, 98] and secondary damage resulting from alcohol-induced changes in fetal cerebral blood flow [41, 46]. Therefore, mitochondria-targeted therapies with ROS scavengers and antioxidants might be a viable therapeutic strategy for the treatment of FAS/FASDs.
The integrity of cortical microvessels is necessary for optimal vascular development. Permeability of the BBB and matrix metalloproteinases (MMP)-induced proteolysis of the neurovascular matrix may influence cortical vascular development. BBB permeability has also been proposed as a possible site of PAE-induced change in cerebral capillaries. However, no documented evidence of prenatal ethanol-induced BBB permeability currently exists. In contrast, MMP-induced proteolysis of the neurovascular matrix may also cause programmed cell death by cell separation from the extracellular matrix [99, 100]. Indeed, glutamate-induced activation of the endothelium protease MMP-9 from pial microvessels of neonates was seen in a mouse model of PAE [101].
As yet, no global mechanism of alcohol-induced impairment to embryonic or fetal brain development has been revealed, and it is very unlikely that a single mechanism can explain the various components of the FASD presentation. In addition, while alcohol is often regarded the principal chemical that causes birth defects (i.e., a teratogen), alcohol’s breakdown products (i.e., its metabolism) may also play a role. For instance, acetaldehyde, a toxin produced by the breakdown of alcohol in the liver and other organs, may accumulate in the fetal brain during prenatal alcohol exposure and may contribute to the development of FASD. Each individual shows a unique mix of alcohol-related consequences, which is influenced by the time, amount, pattern, and length of the mother’s drinking, in addition to hereditary variables. This variation makes it difficult to compare the effects of drinking across individuals.
Conclusion
There is currently no readily available treatment for intentional or unintentional alcohol intake during pregnancy. The inadequate mechanistic knowledge of FASD’s pathogenesis is one of the explanations. More research is required to understand the underlying mechanisms through which alcohol might affect the structure and function of cells. Understanding these multiple mechanisms and seeking to inhibit them may help us to reduce the negative effects of alcohol exposure on embryonic development in the future. Additionally, efforts must be made to improve public awareness of the detrimental consequences of even little alcohol intake during pregnancy.
Author contributions
Study conception and design: PS and WM; Draft manuscript preparation: PS. All authors reviewed the contents and approved the final version of the manuscript.
Funding
Original studies from our laboratory were supported by a grant from the National Institute on Alcohol Abuse and Alcoholism (1 R01 AA027206-01) and funds from the Sanford School of Medicine at the University of South Dakota.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Dejong, K, Olyaei, A, and Lo, JO. Alcohol use in pregnancy. Clin Obstet Gynecol (2019) 62(1):142–55. doi:10.1097/GRF.0000000000000414
2. Finer, LB, and Zolna, MR. Declines in unintended pregnancy in the United States, 2008-2011. N Engl J Med (2016) 374(9):843–52. doi:10.1056/NEJMsa1506575
3. Mattson, SN, Schoenfeld, AM, and Riley, EP. Teratogenic effects of alcohol on brain and behavior. Alcohol Res Health (2001) 25(3):185–91.
4. Caputo, C, Wood, E, and Jabbour, L. Impact of fetal alcohol exposure on body systems: a systematic review. Birth Defects Res C Embryo Today (2016) 108(2):174–80. doi:10.1002/bdrc.21129
5. Thomas, JD, Warren, KR, and Hewitt, BG. Fetal alcohol spectrum disorders: from research to policy. Alcohol Res Health (2010) 33(1-2):118–26.
6. Riley, EP, Infante, MA, and Warren, KR. Fetal alcohol spectrum disorders: an overview. Neuropsychol Rev (2011) 21:73–80. doi:10.1007/s11065-011-9166-x
7. Bukiya, AN, and Dopico, AM. Fetal cerebral circulation as target of maternal alcohol consumption. Alcohol Clin Exp Res (2018) 42(6):1006–18. doi:10.1111/acer.13755
8. Muoio, V, Persson, PB, and Sendeski, MM. The neurovascular unit - concept review. Acta Physiol (2014) 210(4):790–8. doi:10.1111/apha.12250
9. Iadecola, C. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron (2017) 96(1):17–42. doi:10.1016/j.neuron.2017.07.030
10. Bayer, S, Altman, J, Russo, R, and Zhang, X. Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology (1993) 14(1):83–144.
11. Norman, MG, and O’Kusky, JR. The growth and development of microvasculature in human cerebral cortex. J Neuropathol Exp Neurol (1986) 45(3):222–32. doi:10.1097/00005072-198605000-00003
12. Tam, SJ, and Watts, RJ. Connecting vascular and nervous system development: angiogenesis and the bloodbrain barrier. Annu Rev Neurosci (2010) 33:379–408. doi:10.1146/annurev-neuro-060909-152829
13. Walchli, T, Wacker, A, Frei, K, Regli, L, Schwab, ME, Hoerstrup, SP, et al. Wiring the vascular network with neural cues: a CNS perspective. Neuron (2015) 87:271–96. doi:10.1016/j.neuron.2015.06.038
14. Obermeier, B, Daneman, R, and Ransohoff, RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med (2013) 19:1584–96. doi:10.1038/nm.3407
15. Kim, J, Jin, H, Park, JA, Lee, SW, Kim, WJ, Yu, YS, et al. Blood-neural barrier: intercellular communication at glio-vascular interface. J Biochem Mol Biol (2006) 39:339–45. doi:10.5483/bmbrep.2006.39.4.339
16. McConnell, HL, Kersch, CN, Woltjer, RL, and Neuwelt, EA. The translational significance of the neurovascular unit. J Biol Chem (2017) 292:762–70. doi:10.1074/jbc.R116.760215
17. Geudens, I, and Gerhardt, H. Coordinating cell behaviour during blood vessel formation. Development (2011) 138:4569–83. doi:10.1242/dev.062323
18. Siqueira, M, Araujo, APB, Gomes, FCA, and Stipursky, J. Ethanol gestational exposure impairs vascular development and endothelial potential to control BBB-associated astrocyte function in the developing cerebral cortex. Mol Neurobiol (2021) 58(4):1755–68. doi:10.1007/s12035-020-02214-8
19. Bake, S, Tingling, JD, and Miranda, RC. Ethanol exposure during pregnancy persistently attenuates cranially directed blood flow in the developing fetus: evidence from ultrasound imaging in a murine second trimester equivalent model. Alcohol Clin Exp Res (2012) 36(5):748–58. doi:10.1111/j.1530-0277.2011.01676.x
20. Bergers, G, and Benjamin, LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer (2003) 3:401–10. doi:10.1038/nrc1093
21. Carmeliet, P, and Collen, D. Vascular development and disorders: molecular analysis and pathogenic insights. Kidney Int (1998) 53:1519–49. doi:10.1046/j.1523-1755.1998.00936.x
22. Ulrich, F, Carretero-Ortega, J, Menéndez, J, Narvaez, C, Sun, B, Lancaster, E, et al. Reck enables cerebrovascular development by promoting canonical wnt signaling. Development (2016) 143:1055–159. doi:10.1242/dev.136507
23. Wang, X, and Kroenke, CD. Utilization of magnetic resonance imaging in research involving animal models of fetal alcohol spectrum disorders. Alcohol Res (2015) 37:39–51.
24. Williams, JK, Baptista, PM, Daunais, JB, Szeliga, KT, Friedman, DP, and Soker, S. The effects of ethanol consumption on vasculogenesis potential in non human primates. Alcohol Clin Exp Res (2008) 32:155–61. doi:10.1111/j.1530-0277.2007.00558.x
25. Radek, KA, Matthies, AM, Burns, AL, Heinrich, SA, Kovacs, EJ, and Dipietro, LA. Acute ethanol exposure impairs angiogenesis and the proliferative phase of wound healing. Am J Physiol Heart Circ Physiol (2005) 289:H1084–H1090. doi:10.1152/ajpheart.00080.2005
26. Radek, KA, Kovacs, EJ, Gallo, RL, and DiPietro, LA. Acute ethanol exposure disrupts VEGF receptor cell signaling in endothelial cells. Am J Physiol Heart Circ Physiol (2008) 295:H174–H184. doi:10.1152/ajpheart.00699.2007
27. Wang, LL, Yang, AK, He, SM, Liang, J, Zhou, ZW, Li, Y, et al. Identification of molecular targets associated with ethanol toxicity and implications in drug development. Curr Pharm Des (2010) 16:1313–55. doi:10.2174/138161210791034030
28. Alimov, A, Wang, H, Liu, M, Frank, JA, Xu, M, Ou, X, et al. Expression of autophagy and UPR genes in the developing brain during ethanol-sensitive and resistant periods. Metab Brain Dis (2013) 28:667–76. doi:10.1007/s11011-013-9430-2
29. Bisen, S, Kakhniashvili, D, Johnson, DL, and Bukiya, AN. Proteomic analysis of baboon cerebral artery reveals potential pathways of damage by prenatal alcohol exposure. Mol Cel Proteomics (2019) 18(2):294–307. doi:10.1074/mcp.RA118.001047
30. West, JR. Fetal alcohol-induced brain damage and the problem of determining temporal vulnerability: a review. Alcohol Drug Res (1987) 7(5–6):423–41.
31. Popova, EN. Prenatal moderate effects of alcohol on ultrastructure of cortical capillaries in the offspring. Biull Eksp Biol Med (1992) 113:161–4.
32. Kelly, SJ, Mahoney, JC, and West, JR. Changes in brain microvasculature resulting from early postnatal alcohol exposure. Alcohol (1990) 7:43–7. doi:10.1016/0741-8329(90)90059-l
33. Jégou, S, El Ghazi, F, de Lendeu, PK, Marret, S, Laudenbach, V, Uguen, A, et al. Prenatal alcohol exposure affects vasculature development in the neonatal brain. Ann Neurol (2012) 72(6):952–60. doi:10.1002/ana.23699
34. May, PA, and Gossage, JP. Maternal risk factors for fetal alcohol spectrum disorders: not as simple as it might seem. Alcohol Res Health (2011) 34(1):15–26.
35. Maier, SE, and West, JR. Drinking patterns and alcohol-related birth defects. Alcohol Res Health (2001) 25(3):168–74.
36. West, JR, Kelly, SJ, and Pierce, DR. Severity of alcohol-induced deficits in rats during the third trimester equivalent is determined by the pattern of exposure. Alcohol Alcohol Suppl (1987) 1:461–5.
37. Falconer, J, Leonhardt, RA, Agarwal, DP, and Werner Goedde, H. Effect of acute ethanol drinking on alcohol metabolism in subjects with different ADH and ALDH genotypes. Alcohol (1990) 25:413–8. doi:10.1016/0741-8329(90)90025-8
38. Mann, LI, Bhakthavathsalan, A, Liu, M, and Makowski, P. Effect of alcohol on fetal cerebral function and metabolism. Am J Obstet Gynecol (1975) 122:845–51. doi:10.1016/0002-9378(75)90726-7
39. Richardson, BS, Patrick, JE, Bousquet, J, Homan, J, and Brien, JF. Cerebral metabolism in fetal lamb after maternal infusion of ethanol. Am J Physiol (1985) 249:R505–R509. doi:10.1152/ajpregu.1985.249.5.R505
40. Gleason, CA, and Hotchkiss, KJ. Cerebral responses to acute maternal alcohol intoxication in immature fetal sheep. Pediatr Res (1992) 31(6):645–8. doi:10.1203/00006450-199206000-00021
41. Parnell, SE, Ramadoss, J, Delp, MD, Ramsey, MW, Chen, WJ, West, JR, et al. Chronic ethanol increases fetal cerebral blood flow specific to the ethanol-sensitive cerebellum under normoxaemic, hypercapnic and acidaemic conditions: ovine model. Exp Physiol (2007) 92(5):933–43. doi:10.1113/expphysiol.2007.038091
42. Kochunov, P, Castro, C, Davis, DM, Dudley, D, Wey, HY, Purdy, D, et al. Fetal brain during a binge drinking episode: a dynamic susceptibility contrast MRI fetal brain perfusion study. NeuroReport (2010) 21:716–21. doi:10.1097/WNR.0b013e32833b5047
43. North, K, Tobiasz, A, Sullivan, RD, Bursac, Z, Duncan, J, Sullivan, JP, et al. Prenatal alcohol exposure, anesthesia, and fetal loss in baboon model of pregnancy. J Drug Alcohol Res (2018) 7:236064.
44. Simakova, M, Tobiasz, A, Sullivan, RD, Bisen, S, Duncan, J, Sullivan, JP, et al. Gestational age-dependent interplay between endocannabinoid receptors and alcohol in fetal cerebral arteries. J Drug Alcohol Res (2019) 8:236068. doi:10.4303/jdar/236068
45. Tobiasz, AM, Duncan, JR, Bursac, Z, Sullivan, RD, Tate, DL, Dopico, AM, et al. The effect of prenatal alcohol exposure on fetal growth and cardiovascular parameters in a baboon model of pregnancy. Reprod Sci (2018) 25:1116–23. doi:10.1177/1933719117734317
46. Seleverstov, O, Tobiasz, A, Jackson, JS, Sullivan, R, Ma, D, Sullivan, JP, et al. Maternal alcohol exposure during mid-pregnancy dilates fetal cerebral arteries via endocannabinoid receptors. Alcohol (2017) 61:51–61. doi:10.1016/j.alcohol.2017.01.014
47. Mayock, DE, Ness, D, Mondares, RL, and Gleason, CA. Binge alcohol exposure in the second trimester attenuates fetal cerebral blood flow response to hypoxia. J Appl Physiol (2007) 102(3):972–7. doi:10.1152/japplphysiol.00956.2006
48. Mayock, DE, Ngai, AC, Mondares, RL, and Gleason, CA. Effects of binge alcohol exposure in the second trimester on intracerebral arteriolar function in third trimester fetal sheep. Brain Res (2008) 1226:111–5. doi:10.1016/j.brainres.2008.05.077
49. Gleason, CA, Iida, H, Hotchkiss, KJ, Northington, FJ, and Traystman, RJ. Newborn cerebrovascular responses after first trimester moderate maternal ethanol exposure in sheep. Pediatr Res (1997) 42:39–45. doi:10.1203/00006450-199707000-00007
50. Ngai, AC, Mondares, RL, Mayock, DE, and Gleason, CA. Fetal alcohol exposure alters cerebrovascular reactivity to vasoactive intestinal peptide in adult sheep. Neonatology (2008) 93:45–51. doi:10.1159/000105524
51. Turcotte, LA, Aberle, NS, Norby, FL, and Wang, GJ. Influence of prenatal ethanol exposure on vascular contractile response in rat thoracic aorta. Alcohol (2002) 26:75–81. doi:10.1016/s0741-8329(01)00198-7
52. Cananzi, SG, and Mayhan, WG. In utero exposure to alcohol alters reactivity of cerebral arterioles. J Cereb Blood Flow Metab (2017) 39:332–41. doi:10.1177/0271678X17728163
53. Cananzi, SG, and Mayhan, WG. In utero exposure to alcohol impairs reactivity of cerebral arterioles and increases susceptibility of the brain to damage following ischemia/reperfusion in adulthood. Alcohol Clin Exp Res (2019) 43:607–16. doi:10.1111/acer.13979
54. Saha, PS, Kim Sawtelle, KR, Bamberg, BN, Arrick, DM, Watt, MJ, Scholl, JL, et al. Rosiglitazone restores nitric oxide synthase-dependent reactivity of cerebral arterioles in rats exposed to prenatal alcohol. Alcohol Clin Exp Res (2021) 45(7):1359–69. doi:10.1111/acer.14634
55. Saha, PS, Knecht, TM, Arrick, DM, Watt, MJ, Scholl, JL, and Mayhan, WG. Constrictor responses of cerebral resistance arterioles in male and female rats exposed to prenatal alcohol. Physiol Rep (2021) 9(21):e15079. doi:10.14814/phy2.15079
56. Parkington, HC, Kenna, KR, Sozo, F, Coleman, HA, Bocking, A, Brien, JF, et al. Maternal alcohol consumption in pregnancy enhances arterial stiffness and alters vasodilator function that varies between vascular beds in fetal sheep. J Physiol (2014) 592:2591–603. doi:10.1113/jphysiol.2013.262873
57. Daft, PA, Johnston, MC, and Sulik, KK. Abnormal heart and great vessel development following acute ethanol exposure in mice. Teratology (1986) 33:93–104. doi:10.1002/tera.1420330112
58. Coffin, JM, Baroody, S, Schneider, K, and O’Neill, J. Impaired cerebellar learning in children with prenatal alcohol exposure: a comparative study of eyeblink conditioning in children with ADHD and dyslexia. Cortex. (2005) 41:389–98. doi:10.1016/s0010-9452(08)70275-2
59. Bell, SH, Stade, B, Reynolds, JN, Rasmussen, C, Andrew, G, Hwang, PA, et al. The remarkably high prevalence of epilepsy and seizure history in fetal alcohol spectrum disorders. Alcohol Clin Exp Res (2010) 34:1084–9. doi:10.1111/j.1530-0277.2010.01184.x
60. Guerri, C, Bazinet, A, and Riley, EP. Foetal alcohol spectrum disorders and alterations in brain and behaviour. Alcohol Alcohol (2009) 44:108–14. doi:10.1093/alcalc/agn105
61. Heistad, DH, and Lawton, WJ. Pathogenesis of acute hypertensive encephalopathy. In: JL Izzo, and HR Black, editors. Hypertension Primer. 2. Baltimore, MD: Lippincott Williams and Wilkins (1999). p. 186–7.
62. Miller, MW. Effects of alcohol on the generation and migration of cerebral cortical neurons. Science (1986) 233(4770):1308–11. doi:10.1126/science.3749878
63. Bonthius, DJ, and West, JR. Alcohol-induced neuronal loss in developing rats: increased brain damage with binge exposure. Alcohol Clin Exp Res (1990) 14(1):107–18. doi:10.1111/j.1530-0277.1990.tb00455.x
64. Ikonomidou, C, Bittigau, P, Koch, C, Genz, K, Hoerster, F, Felderhoff-Mueser, U, et al. Neurotransmitters and apoptosis in the developing brain. Biochem Pharmacol (2001) 62(4):401–5. doi:10.1016/s0006-2952(01)00696-7
65. Marcussen, BL, Goodlett, CR, Mahoney, JC, and West, JR. Developing rat purkinje cells are more vulnerable to alcohol-induced depletion during differentiation than during neurogenesis. Alcohol (1994) 11(2):147–56. doi:10.1016/0741-8329(94)90056-6
66. Karl, PI, Kwun, R, Slonim, A, and Fisher, SE. Ethanol elevates fetal serum glutamate levels in the rat. Alcohol Clin Exp Res (1995) 19(1):177–81. doi:10.1111/j.1530-0277.1995.tb01488.x
67. Thomas, JD, Weinert, SP, Sharif, S, and Riley, EP. MK-801 administration during ethanol withdrawal in neonatal rat pups attenuates ethanol-induced behavioral deficits. Alcoholism Clin Exp Res (1997) 21(7):1218–25. doi:10.1111/j.1530-0277.1997.tb04441.x
68. Hoffman, PL, Rabe, CS, Moses, F, and Tabakoff, B. N-methyl-D-aspartate receptors and ethanol: inhibition of calcium flux and cyclic GMP production. J Neurochem (1989) 52(6):1937–40. doi:10.1111/j.1471-4159.1989.tb07280.x
69. Hughes, PD, Kim, YN, Randall, PK, and Leslie, SW. Effect of prenatal ethanol exposure on the developmental profile of the NMDA receptor subunits in rat forebrain and hippocampus. Alcoholism Clin Exp Res (1998) 22(6):1255–61. doi:10.1111/j.1530-0277.1998.tb03906.x
70. Jones, PJ, Leichter, J, and Lee, M. Placental blood flow in rats fed alcohol before and during gestation. Life Sci (1981) 29(11):1153–9. doi:10.1016/0024-3205(81)90204-6
71. Mukherjee, AB, and Hodgen, GD. Maternal ethanol exposure induces transient impairment of umbilical circulation and fetal hypoxia in monkeys. Science (1982) 218(4573):700–2. doi:10.1126/science.6890235
72. Abel, EL. Prenatal effects of alcohol. Drug Alcohol Depend. (1984) 14(1):1–10. doi:10.1016/0376-8716(84)90012-7
73. Abel, EL. Prenatal effects of alcohol on growth: a brief overview. Fed Proc (1985) 44(7):2318–22.
74. Kennedy, LA. Changes in the term mouse placenta associated with maternal alcohol consumption and fetal growth deficits. Am J Obstet Gynecol (1984) 149(5):518–22. doi:10.1016/0002-9378(84)90028-0
75. Pennington, SN, Boyd, JW, Kalmus, GW, and Wilson, RW. The molecular mechanism of fetal alcohol syndrome (FAS). I. Ethanol-induced growth suppression. Neurobehav Toxicol Teratol (1983) 5(2):259–62.
76. Ornoy, A. Embryonic oxidative stress as a mechanism of teratogenesis with special emphasis on diabetic embryopathy. Reprod Toxicol (2007) 24(1):31–41. doi:10.1016/j.reprotox.2007.04.004
77. Pollard, I. Neuropharmacology of drugs and alcohol in mother and fetus. Semin Fetal Neonatal Med (2007) 12(2):106–13. doi:10.1016/j.siny.2006.12.001
78. Feng, MJ, Yan, SE, and Yan, QS. Effects of prenatal alcohol exposure on brain-derived neurotrophic factor and its receptor tyrosine kinase B in offspring. Brain Res (2005) 1042(2):125–32. doi:10.1016/j.brainres.2005.02.017
79. Miller, MW. Expression of transforming growth factor-beta in developing rat cerebral cortex: effects of prenatal exposure to ethanol. J Comp Neurol (2003) 460(3):410–24. doi:10.1002/cne.10658
80. Chrissobolis, S, Banfi, B, Sobey, CG, and Faraci, FM. Role of NOX isoforms in angiotensin II induced oxidative stress and endothelial dysfunction in brain. J Appl Physiol (2012) 113:184–91. doi:10.1152/japplphysiol.00455.2012
81. De Silva, TM, and Faraci, FM. Effects of angiotensin II on the cerebral circulation: role of oxidative stress. Front Physiol (2012) 3:484. doi:10.3389/fphys.2012.00484
82. Konior, A, Schramm, A, Czesnikiewicz-Guzik, M, and Guzik, TJ. NADPH oxidases in vascular pathology. Antioxid Redox Signal (2014) 20:2794–814. doi:10.1089/ars.2013.5607
83. Lynch, CM, Kinzenbaw, DA, Chen, X, Zhan, S, Mezzetti, E, Filosa, J, et al. Nox2-derived superoxide contributes to cerebral vascular dysfunction in diet-induced obesity. Stroke (2013) 44(11):3195–201. doi:10.1161/STROKEAHA.113.001366
84. Paravicini, TM, Chrissobolis, S, Drummond, GR, and Sobey, CG. Increased NADPH-oxidase activity and Nox4 expression during chronic hypertension is associated with enhanced cerebral vasodilatation to NADPH In Vivo. Stroke (2004) 35:584–9. doi:10.1161/01.STR.0000112974.37028.58
85. Fukunaga, Y, Itoh, H, Doi, K, Tanaka, T, Yamashita, J, Chun, TH, et al. Thiazolidinediones, peroxisome proliferator-activated receptor gamma agonists, regulate endothelial cell growth and secretion of vasoactive peptides. Atherosclerosis (2001) 158:113–9. doi:10.1016/s0021-9150(01)00430-0
86. Marx, N, Bourcier, T, Sukhova, GK, Libby, P, and Plutzky, J. PPARgamma activation in human endothelial cells increases plasminogen activator inhibitor type-1 expression: PPARgamma as a potential mediator in vascular disease. Arterioscler Thromb Vasc Biol (1999) 19:546–51. doi:10.1161/01.atv.19.3.546
87. Marx, N, Schönbeck, U, Lazar, MA, Libby, P, Plutzky, J, and Schonbeck, U. Peroxisome proliferator-activated receptor gamma activators inhibit gene expression and migration in human vascular smooth muscle cells. Circ Res (1998) 83:1097–103. doi:10.1161/01.res.83.11.1097
88. Grant, S, Lutz, EM, McPhaden, AR, and Wadsworth, RM. Location and function of VPAC1, VPAC2 and NPR-C receptors in VIP-induced vasodilation of porcine basilar arteries. J Cereb Blood Flow Metab (2006) 26:58–67. doi:10.1038/sj.jcbfm.9600163
89. Gaw, AJ, Aberdeen, J, Humphrey, PP, Wadsworth, RM, and Burnstock, G. Relaxation of sheep cerebral arteries by vasoactive intestinal polypeptide and neurogenic stimulation: inhibition by L -NG-monomethyl arginine in endothelium-denuded vessels. Br J Pharmacol (1991) 102:567–72. doi:10.1111/j.1476-5381.1991.tb12213.x
90. Wei, EP, Kontos, HA, and Said, SI. Mechanism of action of vasoactive intestinal polypeptide on cerebral arterioles. Am J Physiol (1980) 239:H765–H768. doi:10.1152/ajpheart.1980.239.6.H765
91. Dacey, RG, Bassett, JE, and Takayasu, M. Vasomotor responses of rat intracerebral arterioles to vasoactive intestinal peptide, substance P, neuropeptide Y, and bradykinin. J Cereb Blood Flow Metab (1988) 8:254–61. doi:10.1038/jcbfm.1988.56
92. Spong, CY, Auth, J, Vink, J, Goodwin, K, Abebe, DT, Hill, JM, et al. Vasoactive intestinal peptide mRNA and immunoreactivity are decreased in fetal alcohol syndrome model. Regul Pept (2002) 108:143–7. doi:10.1016/s0167-0115(02)00104-0
93. Olsson, AK, Dimberg, A, Kreuger, J, and Claesson-Welsh, L. VEGF receptor signalling: in control of vascular function. Nat Rev Mol Cel Biol. (2006) 7:359–71. doi:10.1038/nrm1911
94. Girault, V, Gilard, V, Marguet, F, Lesueur, C, Hauchecorne, M, Ramdani, Y, et al. Prenatal alcohol exposure impairs autophagy in neonatal brain cortical microvessels. Cell Death Dis. (2017) 8:e2610. doi:10.1038/cddis.2017.29
95. Semple, BD, Blomgren, K, Gimlin, K, Ferriero, DM, and Noble-Haeusslein, LJ. Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol (2013) 106–107:1–16. doi:10.1016/j.pneurobio.2013.04.001
96. Chen, G, Ke, Z, Xu, M, Liao, M, Wang, X, Qi, Y, et al. Autophagy is a protective response to ethanol neurotoxicity. Autophagy (2012) 8:1577–89. doi:10.4161/auto.21376
97. Bukiya, AN. Fetal cerebral artery mitochondrion as target of prenatal alcohol exposure. Int J Environ Res Public Health (2019) 16(9):1586. doi:10.3390/ijerph16091586
98. Manzo-Avalos, S, and Saavedra-Molina, A. Cellular and mitochondrial effects of alcohol consumption. Int J Environ Res Public Health (2010) 7:4281–304. doi:10.3390/ijerph7124281
99. Tagaya, M, Haring, HP, Stuiver, I, Wagner, S, Abumiya, T, Lucero, J, et al. Rapid loss of microvascular integrin expression during focal brain ischemia reflects neuron injury. J Cereb Blood Flow Metab (2001) 21(7):835–46. doi:10.1097/00004647-200107000-00009
100. Chen, ZL, and Strickland, S. Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell (1997) 91(7):917–25. doi:10.1016/s0092-8674(00)80483-3
Keywords: brain, cerebral resistance arterioles, prenatal alcohol exposure, FASD, cerebral blood flow
Citation: Saha PS and Mayhan WG (2022) Prenatal exposure to alcohol: mechanisms of cerebral vascular damage and lifelong consequences. Adv. Drug. Alco. Res. 2:10818. doi: 10.3389/adar.2022.10818
Received: 07 August 2022; Accepted: 01 November 2022;
Published: 21 November 2022.
Edited by:
Declan Ali, University of Alberta, CanadaReviewed by:
Anna Bukiya, University of Tennessee Health Science Center (UTHSC), United StatesAmy Gardiner, University of New Mexico, United States
Copyright © 2022 Saha and Mayhan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Partha S. Saha, cGFydGhhLnNhaGFAdXNkLmVkdQ==