 Klaudia Babišová1*
Klaudia Babišová1* Patrik Krumpolec1Dominik Hadžega1
Patrik Krumpolec1Dominik Hadžega1 Peter Sabaka2Peter Jackuliak3Gabriel Minárik1Michaela Hýblová1
Peter Sabaka2Peter Jackuliak3Gabriel Minárik1Michaela Hýblová1- 1Medirex Group Academy n.o., Nitra, Slovakia
- 2Department of Infectology and Geographical Medicine, Faculty of Medicine, Comenius University in Bratislava, Bratislava, Slovakia
- 35th Department of Internal Medicine, Comenius University Faculty of Medicine, University Hospital Bratislava, Bratislava, Slovakia
Since its outbreak in late 2019, the SARS-CoV-2 virus has been the main subject of interest for a number of studies. Clinical manifestations are ranging from asymptomatic to mild and severe. Major risk factors for developing severe COVID-19 are age and comorbidities, although younger people suffer from severe COVID-19 as well. One of the explanations for why can be the composition of respiratory tract microbiota. In this article, we review studies linking respiratory tract microbiome and its changes during COVID-19 infection. The respiratory tract microbiome helps shape immunity and it is assumed that it can affect the outcome of several viral infections. Several studies show differences in the microbial composition of the respiratory tract between COVID-19 patients and healthy individuals. The diversity of the respiratory tract microbiome is reduced with increasing severity of COVID-19.
SARS-CoV-2
In late December 2019, several patients with symptoms of novel pneumonia were reported to health authorities in Wuhan, Hubei Province, China (Zhou et al., 2020). The majority of the initial patients were associated with the Huanan seafood and wildlife market in Wuhan City. Through retrospective analysis, they were able to identify a patient whose symptoms began as early as 1 December (Zhang and Holmes, 2020). Isolation of viral RNA and next-generation sequencing identified this viral pathogen as a novel coronavirus (Zhou et al., 2020). The International Committee on Taxonomy of Viruses named this novel coronavirus Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) (Gorbalenya et al., 2020).
SARS-CoV-2 belongs to the viral family Coronaviridae. Members of the family Coronaviridae are single-stranded positive-sense RNA viruses up to 32 kb in length. In addition, this family can be divided into two subfamilies: Coronavirinae and Torovirinae. Subfamily Coronavirinae is divided into four genera: Alphacoronavirus, Betacoronavirus, Gammacoronavirus, Deltacoronavirus and subfamily Torovirinae, which consists of the genus Torovirus (Payne, 2017). Alphacoronavirus and Betacoronavirus can generally infect mammals, whereas Gammacoronavirus and Deltacoronavirus mainly infect birds (Ludwig and Zarbock, 2020). In humans, they are the cause of mild to severe respiratory infections. By 2019, in the 21st century, humanity experienced two spreads of novel coronaviruses: severe acute respiratory syndrome (SARS) in 2002 and Middle East respiratory syndrome (MERS) in 2012 (de Wit et al., 2016; Hu et al., 2020). SARS occurred in China in 2002, sickening nearly 8,000 people and killing 774. The natural reservoir for SARS-CoV was Chinese horseshoe bats (Lau et al., 2005), but transmission to humans occurred via intermediate hosts, civets (Kan et al., 2005). MERS-CoV emerged in Saudi Arabia in 2012 and infected approximately 2,500 people with a mortality rate of 30% (Lu et al., 2020). As in the case of SARS, the MERS-CoV virus persists in nature in bats and as an intermediate host serve dromedaries (Sabir et al., 2016). A study by Zhou et al. (2020) showed that SARS-CoV-2 is 96.2% identical to bat coronavirus at the whole genome level, further supporting the theory that bats are natural reservoirs for coronaviruses. It is unknown whether SARS-CoV-2 was transmitted directly from a bat to a human or whether there is another intermediate host. Recent studies speculated that intermediate hosts could be rodents (Huang et al., 2021), pangolins (Liu et al., 2020) and raccoon dogs (Wang et al., 2022), but there could be multiple intermediate hosts.
Next-generation sequencing revealed 79% nucleotide-level identity between SARS-CoV-1 and SARS-CoV-2 and 50% between SARS-CoV-2 and MERS-CoV (Lu et al., 2020). The genome of SARS-CoV-2 resembles that of a typical coronavirus genome and contains at least nine open reading frames and four structural proteins with the gene sequence 5′-ORF1ab-S-E-M-N-3′ (S-spike, E-envelope, M-membrane, N-nucleocapsid protein) (Figure 1) (Li et al., 2020). ORF1a and ORF1b are two overlapping ORFs encoding polyproteins pp1a and pp1ab, which are cleaved into 16 non-structural proteins (NSP) (Ziebuhr and Siddell, 1999; Abu Turab Naqvi et al., 2020; Dutartre et al., 2020; Yadav et al., 2021). These NSPs (1-16) are responsible for viral replication (Yadav et al., 2021). The spike (S) protein ensures the entry of the virus into the target cell. The S protein consists of two subunits, the N-terminal S1 subunit and the C-terminal S2 subunit. The S1 unit recognizes and binds to the cell surface receptor angiotensin-converting enzyme-2 (ACE-2), the same one that binds the spike protein of SARS-CoV-1, followed by fusion of viral and host cellular membrane. Binding to the ACE-2 receptor requires S-protein priming by the host cellular serine TMPRSS2 protease. This priming involves cleavage at the S1/S2 furin cleavage site, making the closed form of S1 open and accessible to the ACE-2 receptor (Benton et al., 2020; Hoffmann et al., 2020; Guruprasad, 2021). ACE-2 is expressed in epithelial tissues that form protective barriers. ACE-2 is localized in many cells and tissues such as the heart, kidneys, intestinal tract, gallbladder, testis, and most importantly, respiratory tract epithelium in the nose, mouth, and lungs. In the lungs, it is strongly expressed in the pneumocytes in the alveoli (Hikmet et al., 2020; Wang et al., 2020). Proteins M and E form the viral envelope, while protein N protects viral RNA (Kirtipal et al., 2020).
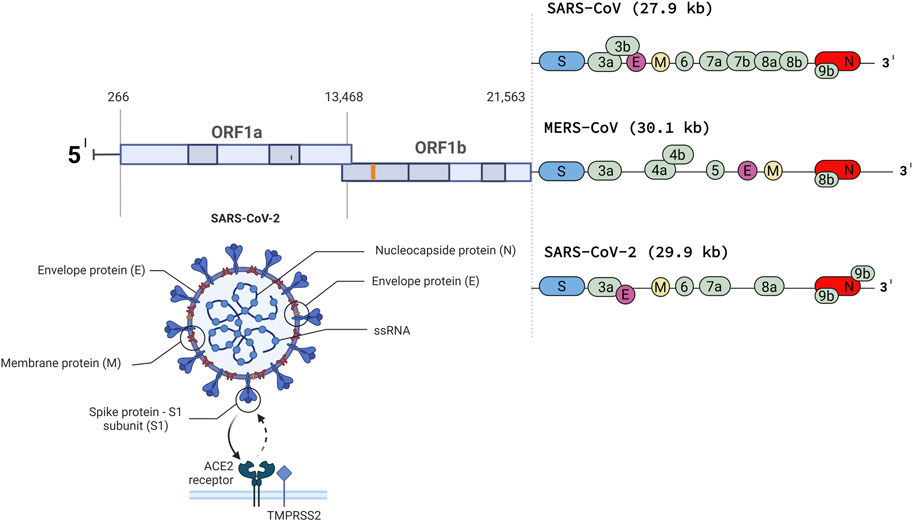
FIGURE 1. The genome of SARS-CoV, MERS-CoV and SARSCoV- 2 (Created with BioRender.com).
SARS-CoV-2 has various manifestations in patients, from mild to severe. Unlike SARS and MERS, the mortality rate of SARS-CoV-2 is significantly lower, with most patients having mild symptoms or being asymptomatic. Symptoms vary from patient to patient and depend on various factors such as age, gender, blood type, and other comorbidities (diabetes, hypertension, cardiovascular disease, etc.) (Huang et al., 2020). The most common symptoms are fever, dry cough, fatigue, sputum production, and loss of taste and smell. Less common symptoms include headache, sore throat, diarrhea, chills, nausea, and vomiting (Hu et al., 2020; Ludwig and Zarbock, 2020).
Respiratory tract
The respiratory tract (RT) is a part of the respiratory system, whose main function is the exchange of oxygen and carbon dioxide. It is structurally divided into the upper respiratory tract (URT) and the lower respiratory tract (LRT). The UTR consists of the nostrils, nasopharynx, oropharynx, and the portion of the larynx above the vocal cords. The LRT, on the other hand, includes the portion of the larynx below the vocal cords, the trachea, bronchi, bronchioles, and lungs with alveoli (Robinson and Furlow, 2007). The entire surface is covered by bacterial communities, with the UTR having the highest density of bacterial communities. Different anatomical structures of the respiratory tract contain specialized bacterial communities. The human microbiome is the collection of all microorganisms that live in association with the human body. The microbiome includes eukaryotes, archaea, bacteria, and viruses (https://www.hmpdacc.org/overview/). The most studied parts of the microbiome are bacteria, commensal or pathogenic. The RT microbiota is thought to play a role in respiratory development and in shaping local immunity (Olszak et al., 2012). During bacterial or viral infection of the human respiratory tract, the first step for these pathogens is usually colonization of the UTR before causing infection of the UTR and LTR. The process of inhibiting this first step is also referred to as colonization resistance and its mechanisms involve competition for the attachment site, nutrition, or production of antibacterial peptides (Stadio et al., 2020).
Composition of RT microbiota
Colonization of the URT begins at birth and is influenced by the mode of delivery. During vaginal delivery, the child is exposed to the maternal vaginal microbiome, and during cesarean delivery, the infant is exposed to the maternal skin and environmental microbiome. During the first week of life, the nasopharynx is colonized by Staphylococcus aureus regardless of the mode of delivery. Although S. aureus is primarily known as a pathogenic bacterium, it may be an important commensal bacterium in the nasopharynx during early life. Wang et al. (2013) showed that S. aureus significantly attenuates influenza-mediated immune injury of the lung in mice by inducing alveolar macrophages. In later weeks, the abundance of S. aureus decreases and species such as Corynebacterium, Dolosigranulum, and Moraxella increase. Increased abundance of these three species is important for the healthy development of the nasopharyngeal microbiota in later stages of life. In the first months of life, infants born by cesarean section exhibited a variable microbial profile and a loss of abundance of Corynebacterium and Dolosigranulum, resulting in an increase in respiratory infections. The abundance of S. aureus is not significantly reduced, while Prevotella, Veillonela, and Porphyromonas begin to emerge (Bosch et al., 2017). In contrast, infants born naturally and breastfed showed a higher abundance of beneficial bacteria (Bosch et al., 2016). This could also be due to the transfer of beneficial microbiota, such as Lactobacillus and Bifidobacterium, in milk during breastfeeding (Biesbroek et al., 2014). Breastfed children also have a higher abundance of Corynebacterium and Dolosigranulum and a lower incidence of respiratory diseases compared to formula-fed children. The microbiota of children can be influenced by many different aspects, such as the type of birth and feeding mentioned above, presence of siblings, previous infections, use of antibiotics, vaccinations, season, exposure to different environments (home, kindergarten, park), etc. In the first year of life, exposure to microbial communities is crucial for the formation of the immune system (Shukla et al., 2017).
The human body is colonized by five phyla of bacteria: Actinobacteria, Bacteroidetes, Firmicutes, Fusobacteria, and Proteobacteria (Ibironke et al., 2020). In healthy adults, the nasal cavity microbiome consists of bacteria associated with the skin, mainly Actinobacteria with the most abundant families being Corynebacteriaceae and Propionibacteriaceae, followed by Firmicutes and Proteobacteria (Bassis et al., 2014). The nasopharynx differs from the oral cavity in terms of bacterial niche composition. In spring, the dominant phyla in the nasopharynx are Bacteroidetes, and in fall, Proteobacteria and Firmicutes. At the genus level, Moraxella is the most widespread (Bogaert et al., 2011). Firmicutes, Proteobacteria, and Bacteroidetes are represented in the oropharynx. The oropharynx has a greater bacterial diversity compared to the nasopharynx (Charlson et al., 2010). Commonly known pathogenic bacteria such as Pneumococcus, Haemophilus influenzae, and Neisseria meningitidis can be considered commensal bacteria of the URT (Table 1). Despite the dogma that the lung is sterile, it has quite a dynamic microbiome. The microbiome of the lung is similar to the microbiome of the nasopharynx in terms of ecological composition (Dickson et al., 2014). The most dominant phyla in the lung are Firmicutes and Bacteroidetes with a low biomass of Prevotella, Veillonella, and Streptococcus. Bacteria enter the lungs mainly through the UTR (Cui et al., 2014). However, Tropheryma whipplei was found in the lung as part of the microbiota, but not in the nasopharynx or oropharynx (Charlson et al., 2011). When comparing the microbiota of middle-aged people and seniors, seniors had the lowest abundance, especially in the anterior nares. In the anterior nares of elderly people, the relative abundance of Propionibacterium and Corynebacterium decreases and Streptococcus increases. The genus Streptococcus was depleted in the oropharynx. The anterior nares are barriers between the external environment and the respiratory tract, so changes in their microbiome may be one of the causes of the higher incidence of respiratory infections in seniors (Whelan et al., 2014).
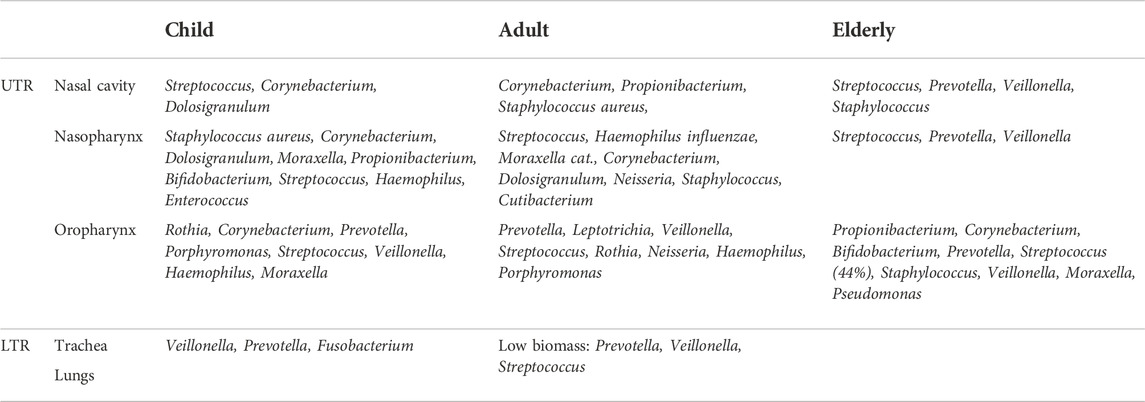
TABLE 1. Composition of microbial communities in various anatomical structures in different stages of life.
The nasal mucosa and its microbiota are the first to come into contact with the infection. Viral infection of the respiratory tract alters the composition of the microbiota in RT, mainly through the decrease of alpha diversity (the average species diversity within a given area) and the loss of beneficial microbiota, mainly anaerobes and the genus Prevotella. On the other hand, pathogenic bacteria such as Staphylococcus aureus, Haemophilus influenzae, Streptococcus pneumoniae, Corynebacterium propinquum and Moraxella catarrhalis are enriched (Edouard et al., 2018). These changes contribute to the ease of shifting infection from the UTR to LTR and allow bacterial pathogens to translocate as well, leading to secondary bacterial infection (Cyprian et al., 2021) and drastically altering the mortality of viral infections (Gupta et al., 2008).
The microbiota can reduce pathogens by alerting the host immune system or directly inhibiting or killing pathogens. A study by Larsen et al. (2012) showed that Prevotella can reduce Haemophilus-induced production of proinflammatory cytokines in dendritic cells by ∼50% (Larsen et al., 2012). The signaling pathway, by which the commensal activates the host immune response is unclear, but it activates cells of host immunity, for example, macrophages, mucosa-associated invariant T cells, group 3 innate lymphoid cells, and natural killer cells. Murine studies have shown that mice with a lowered abundance of microbiota reduced by antibiotic use had lower expression of macrophage-induced viral genes upon influenza virus infection than the control group (Abt et al., 2012). In addition, Staphylococcus epidermidis activates the production of interferon in the nasal epithelium, increasing resistance to influenza A in mice (Kim et al., 2019). Two main ways in which bacteria compete with each other are exploitation and interference. In exploitation, bacteria extract nutrients from the environment. Corynebacterium produces siderophores and inhibits Staphylococcus growth due to decreased bioavailability of iron as a result of the chelation of iron by siderophores in vitro (Stubbendieck et al., 2019). In contrast, competing bacteria produce molecules during interference that inhibit growth or kill competitors. Bacteria can produce bacteriocins and ribosomally synthesized antimicrobial peptides that can inhibit growth or kill other bacteria, but do not harm the bacteria themselves through specific immunity proteins (Benítez-Chao et al., 2021; Soltani et al., 2021). Staphylococcus salivarius produces bacteriocins and thus inhibits Staphylococcus pneumoniae (Santagati et al., 2012). Other mechanisms of interference include biofilm disruption, enzyme secretion, hydrogen peroxide production, and virulence gene downregulation (Khan et al., 2019).
COVID-19 and respiratory microbiome
As mentioned earlier, the microbiota of the respiratory tract plays a crucial role in maintaining respiratory health. Multiple studies have shown that the composition of respiratory microbiota can affect susceptibility to respiratory infections including COVID-19. Understanding interactions between the host, virus and respiratory microbiome may provide insight into new potential therapeutic targets in the prevention and treatment of those infections. SARS-CoV-2 enters the human body primarily through the respiratory tract, oral cavity, and nose. The microbiome is the first entity to confront infection. It plays an important role in stimulating the immune system and protecting against pathogens. When microbiome homeostasis is disturbed (microbial dysbiosis), pathogens overgrow and colonize the respiratory tract, eventually leading to infection of the LTR. In samples from patients with severe COVID-19 bacterial alpha diversity decreased. Furthermore, alpha diversity is reduced in a severity-dependent fashion—the lower diversity, the higher severity. As with most infections, the trend of beneficial bacteria decreasing and pathogens increasing can be seen in SARS-CoV-2 as well.
The severity of COVID-19 is related to age and comorbidities. Since the elderly have low diversity of the microbiome in RT, this may be the reason for their high susceptibility to SARS-CoV-2 and their higher probability of ending up in the intensive care unit (ICU). At the same time, young patients with no apparent risk factors suffer complications and end up in the ICU on automatic ventilation. The role of the microbiome in the host immune response suggests that a healthy microbiome may positively influence the progression of COVID-19. On the other hand, dysbiosis may lead to a higher rate of severe cases in individuals without obvious risk factors. The URT microbiome may impact the symptoms experienced during SARS-CoV-2 infection by modulating the host immune response. For example, the ACE-2 receptor, as an entry site for SARS-CoV-2, is stimulated by interferon and therefore may be affected by the microbiome. Respiratory tract viral infections may predispose patients to bacterial superinfections. One of the most important mechanisms is the structural and functional disruption of the mucosal epithelium (Bakaletz, 1995). In previous studies, coronavirus enhanced streptococcal attachment to epithelial cells, resulting in pulmonary infections (Golda et al., 2011). Patients with additional coinfection have a higher risk of severe COVID-19 and a higher mortality rate. In the study by Zhou et al., 50% of COVID-19 patients had a secondary bacterial infection. The three most common bacteria in patients’ lungs were Klebsiella pneumoniae (9.9%), Streptococcus pneumoniae (8.2%), and Staphylococcus aureus (7.7%) (Tang et al., 2022).
In a study in which Gupta et al., 2022 analyzed microbial composition at the bacterial strain level, there was an increased abundance of Proteobacteria and decreased abundance of Bacteroidetes. In mice, Bacteroidetes can downregulate ACE-2 receptor expression, this reduction can promote severe SARS-CoV-2 infection. COVID-19 patients’ nasal microbiome is enriched in pathogenic bacteria, Acinetobacter, Rothia, Moraxella, Haemophilus, Stenotrophomonas, and Pseudomonas (Rhoades et al., 2021; Gupta et al., 2022).
In the nasopharynx, Corynebacterium and Dolosigranulum are negatively correlated with COVID-19 severity (Smith et al., 2021). Corynebacterium and Dolosigranulum are among the most important commensal bacteria in healthy RT (Shilts et al., 2022). Corynebacterium accolens can inhibit the growth of Staphylococcus aureus and Staphylococcus pneumoniae (Bomar et al., 2016). In addition, COVID-19 patients show a significant reduction in abundance of Proteobacteria (Kolhe et al., 2021) and Fusobacteria at the phylum level compared to controls (Nardelli et al., 2021). Leptotrichia and Haemophilus are significantly reduced and species such as Streptococcus, Prevotella and Campylobacter are enriched (Nardelli et al., 2021; Xiong et al., 2021). Overexpressed Prevotella proteins can increase COVID-19 severity by interacting with NF-kB (Khan and Khan, 2020; Ventero et al., 2021). The study showed that two Streptococcus strains, S. suis and S. agalactiae, can stimulate the expression of ACE-2 in VERO cells (Xiong et al., 2021), which could be another reason for severe COVID-19. Campylobacter is known to cause inflammation and diarrhea, one of the symptoms of COVID-19. In COVID-19 patients, several pathways were significantly downregulated in the nasopharyngeal metabolome, including platelet activation pathway (Liu et al., 2021).
The bacterial communities in the oropharynx show a higher relative abundance of Streptococcus, Gemella, Haemophilus, and Neisseria, and a lower relative abundance of the phylum Proteobacteria (Liu et al., 2021). In addition, a study by Shi et al. presented three predominant bacteria in the oropharynx in COVID-19 patients with mild symptoms: Streptococcus, Veillonella, and Haemophilus (∼60%). In addition, Campylobacter (including C. rectus and C. fetus) and Roseburia were limited at the genus level in patients with a higher viral load of SARS-CoV-2 (Shi et al., 2022). SARS-CoV-2 intensive care patients showed complete depletion of Clostridium and Bifidobacterium. In addition, the Pseudomonadaceae family was found exclusively in ICU patients compared with hospitalized patients and healthy controls (Rueca et al., 2021). Members of the Pseudomonadaceae family are known to cause a pathological condition and reduce microbial diversity (Borges et al., 2018) In their study Ren et al. identified Streptococcus as the most enriched microbiome at the time of admission in the oropharynx of a patient recovered from COVID-19 (Ren et al., 2021). In the oropharynx of COVID-19 patients, specific amino acid metabolic pathways were enriched compared to healthy control and influenza patients. Tyrosine, phenylalanine, beta-alanine, phosphonate, and phosphinate metabolic pathways were those enriched in COVID-19 patients, suggesting that specific amino acids are metabolized by the microbiome in the oropharynx of COVID-19 patients (Ma et al., 2021). The amino acid imbalance was the reason for increased intestinal inflammation via ACE2-dependent changes in epithelial immunity (Hashimoto et al., 2012). Moreover, the oropharyngeal microbiome of COVID-19 and flu patients showed relatively depleted nucleotide metabolism, replication and repair, and greatly depleted membrane transport and cell motility. These findings suggest a lowered ability to process genetic information and transport ions, lipids, sterols, peptides, proteins and carbohydrates (Ma et al., 2021).
In the lungs of deceased patients with COVID-19, the most common bacteria were Actinobacter (Fan et al., 2020). The difference in lung microbiome composition between healthy individuals and patients with COVID-19 suggests lung dysbiosis during infection with SARS-CoV-2. Intubated patients have lower microbiome diversity, respiratory pathogens such as Staphylococcus, Klebsiella, and Stenotrophemonas and typical bacterial communities for UTR such as Corynebacterium and Prevotella are present. Prevotella is known for its ability to promote viral infections by interacting with NF-κB (Bertelsen et al., 2020). Some patients had a high abundance of Enterococcus, which causes bloodstream infection in critically ill COVID-19 patients (Merenstein et al., 2021). Patients on automatic ventilation have elevated levels of Serratia, Streptococcus, Enterobacter, Veillonella, Prevotella, and Rothia (Feehan et al., 2021), but it is not known whether this is directly from COVID-19 or from intubation and pulmonary ventilation. Serratia marcescens is known to cause pneumonia (Goldstein et al., 1982). A study by Haiminen et al. (2021) focused on altered lung microbiome pathways; patients with COVID-19 had downregulated pathways related to glycan biosynthesis and metabolism and lipid metabolism and increased carbohydrate metabolism (Haiminen et al., 2021).
During infection, the LRT produces mucus by activating various immune cells (Li and Tang, 2021). The presence of mucus in LTR promotes the growth of anaerobic bacteria and, at the same time, reduces the growth of aerobic bacteria. This supports the findings that Staphylococcus, Prevotella, and Peptostreptococcus are increased in COVID-19 patients in critical condition (Smith et al., 2021). Prevotella and Peptostreptococcus are anaerobic bacteria (Brook, 2001), Staphylococcus grows best in the presence of oxygen but can live in anaerobic conditions as well.
Changes in UTR microbiota during SARS-CoV-2 infection are extensively studied, but little is known about changes in metabolome in COVID-19 patients. The main target of metabolomic studies is the focus on the gut metabolome. Recent studies suggest that changes in the respiratory tract microbiome and its metabolites may be a contributing factor to the severity of COVID-19. A study by Clausen et al. (2020) shows that heparan sulfate (HS) is needed for the binding of SARS-CoV-2 to the ACE2 receptor. SARS-CoV-2 infection and binding of the virus to the receptor can be eliminated by the removal of HS from the cell surface (Clausen et al., 2020). Bacteria have co-localized set of genes, called polysaccharide utilization locus (PUL), that are responsible for the degradation of complex carbohydrates, including HS (Terrapon et al., 2015; Martino et al., 2020). PULs are prevalent in Bacteroidetes, the incidence of which is lower in patients with COVID-19.
In a study by Liu et al. (2008) in a macrophage-derived cell line, proinflammatory cytokine production was induced by histone-like DNA binding protein produced by Streptococcus intermedius. Therefore, elevated levels of Streptococcus can be one of the reasons for higher inflammation in COVID-19 patients.
Another metabolite produced by respiratory microbiota such as Lactobacillus spp. is lactic acid (Abedi et al., 2017). Lactic acid can inhibit the growth of pathogenic bacteria by maintaining low pH in the respiratory tract and enhancing the activity of immune cells. Lactic acid-producing bacteria can regulate the population of natural killer cells and thereby inhibiting the influenza A virus (Miyazaki, 2017). Natural killer cells are one of the first components of immunity to respond also to SARS-CoV-2 infection and are important in controlling the first stages of the infection.
Nitric oxide (NO) has an important role in modulating immune response through various mechanisms. NO is produced by cells of innate immunity (macrophages, neutrophils and natural killer cells). Macrophages activated by pathogen release NO, therefore, which inhibits the replication of the pathogen (Tripathi et al., 2007; Wink et al., 2011; Rosier et al., 2020). In their study, Åkerström et al. (2005) infected cells with the SARS-CoV virus and then the cells were treated with different concentrations of the NO donors S-nitroso-N-acetylpenicillamine (SNAP). SNAP inhibited viral replication of SARS-CoV in a dose-dependent manner. When SNAP was used on cells infected with SARS-CoV-2, the results show the same SNAP dose-dependent reduction of SARS-CoV-2 viral RNA copy numbers. The same was seen when observing the correlation between SNAP and the development of viral cytopathic effect. However, if noninfected control cells were treated with 400 µM SNAP, SNAP exhibited a cytotoxic effect. However, cells treated with 200 µM SNAP showed no decrease in viability (Akaberi et al., 2020). Reactive nitrogen species (RNS) can be formed if NO interacts with reactive oxygen species (ROS). When cells are exposed to oxidative stress, RNS may have a cytotoxic effect (Ricciardolo et al., 2004). Several bacteria can produce NO by denitrification of nitrate to nitrite and to NO (Zumft, 1993). Human saliva contains nitrate and oral microbiota (Veillonella spp., Staphylococcus aureus and Staphylococcus epidermidis, Nocardia spp., Corynebacterium spp., Neisseria spp., Actinomyces spp., Haemophilus spp.) is responsible for its conversion to nitrite (Macknight et al., 1997; Hyde et al., 2014).
Conclusion
Respiratory microbiota is a powerful tool in the defense system against viruses. Recent studies show different RT microbial compositions in patients infected with coronavirus SARS-CoV-2, for example, reduced or increased prevalence of bacteria involved in mechanisms that are able to change the progression of COVID-19 disease. Results from these studies are ambiguous, with conflicting conclusions. The composition of the microbiota can be influenced by many factors, including the method of sampling. COVID-19 outbreak as a global pandemic is studied around the globe, but the microbiota of healthy individuals is altered between individuals from different environments. So, studies from different countries with different conditions will not be coherent. Moreover, some studies did not conduct a thorough background investigation, so they do not have information about previous infections or antibiotic use prior to the infection with SARS-CoV-2. Antibiotic use changes the microbiome 4 weeks after administration of the last dose. The sample size was small in most studies as well.
However, the most important finding is that the microbiome is altered in patients infected with COVID-19. Changes in bacterial composition may be the cause of a wide variety of COVID-19 manifestations. It is unknown whether this change contributes to SARS-CoV-2 infection or is the consequence of COVID-19. Nevertheless, alpha diversity is reduced as well as the number of commensal bacteria, while the relative abundance of pathogens increases. Pathogens that are not fully controlled by commensals exacerbate the manifestation of COVID-19.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by the OP Integrated Infrastructure for the project: Serious Diseases of Civilization and COVID-19, ITMS: 313011AVH7, co-financed by the European Regional Development Fund.
Conflict of interest
The authors declare that the article was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abedi, E., Mohammad, S., and Hashemi, B. (2017). Lactic acid production - producing microorganisms and substrates sources-state of art. Heliyon 6, e04974. doi:10.1016/j.heliyon.2020.e04974
Abt, M. C., Osborne, L. C., Monticelli, L. A., Doering, T. A., Alenghat, T., Sonnenberg, G. F., et al. (2012). Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 37 (1), 158–170. doi:10.1016/j.immuni.2012.04.011
Abu Turab Naqvi, A., Fatima, K., Mohammad, T., Fatima, U., Singh, I. K., Singh, A., et al. (2020). Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta Mol. Basis Dis. 1866 (10), 165878. doi:10.1016/j.bbadis.2020.165878
Akaberi, D., Krambrich, J., Ling, J., Luni, C., Hedenstierna, G., Järhult, J. D., et al. (2020). Mitigation of the replication of SARS-CoV-2 by nitric oxide in vitro. Redox Biol. 37, 101734. doi:10.1016/J.REDOX.2020.101734
Åkerström, S., Mousavi-Jazi, M., Klingström, J., Leijon, M., Lundkvist, Å., and Mirazimi, A. (2005). Nitric oxide inhibits the replication cycle of severe acute respiratory syndrome coronavirus. J. Virol. 79 (3), 1966–1969. doi:10.1128/JVI.79.3.1966-1969.2005
Bakaletz, L. (1995). Viral potentiation of bacterial superinfection of the respiratory tract. Trends Microbiol. 3 (3), 110–114. doi:10.1016/S0966-842X(00)88892-7
Bassis, C. M., Tang, A. L., Young, V. B., and Pynnonen, M. A. (2014). The nasal cavity microbiota of healthy adults. Microbiome 2 (1), 27. doi:10.1186/2049-2618-2-27
Benítez-Chao, D. F., León-Buitimea, A., Lerma-Escalera, J. A., and Morones-Ramírez, J. R. (2021). Bacteriocins: An overview of antimicrobial, toxicity, and biosafety assessment by in vivo models. Front Microbiol. 12, 630695. doi:10.3389/fmicb.2021.630695
Benton, D. J., Wrobel, A. G., Xu, P., Roustan, C., Martin, S. R., Rosenthal, P. B., et al. (2020). Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 588 (7837), 327–330. doi:10.1038/s41586-020-2772-0
Bertelsen, A., Elborn, S. J., and Schock Id, B. C. (2020). Toll like Receptor signalling by Prevotella histicola activates alternative NF-κB signalling in Cystic Fibrosis bronchial epithelial cells compared to P. aeruginosa. PLoS One 15 (10), e0235803. doi:10.1371/journal.pone.0235803
Biesbroek, G., Bosch, A. A. T. M., Wang, X., Keijser, B. J. F., Veenhoven, R. H., Sanders, E. A. M., et al. (2014). The impact of breastfeeding on nasopharyngeal microbial communities in infants. Am. J. Respir. Crit. Care Med. 190 (3), 298–308. doi:10.1164/rccm.201401-0073OC
Bogaert, D., Keijser, B., Huse, S., Rossen, J., Veenhoven, R., van Gils, E., et al. (2011). Variability and diversity of nasopharyngeal microbiota in children: A metagenomic analysis. PLoS ONE 6 (2), 17035. doi:10.1371/journal.pone.0017035
Bomar, L., Brugger, S. D., Yost, B. H., Davies, S. S., Lemon, K. P., and Huffnagle, G. B. (2016). Corynebacterium accolens releases antipneumococcal free fatty acids from human nostril and skin surface triacylglycerols. mBio 7, e01725. doi:10.1128/mBio.01725-15
Borges, L. G. D. A., Giongo, A., Pereira, L. de M., Trindade, F. J., Gregianini, T. S., Campos, F. S., et al. (2018). Comparison of the nasopharynx microbiome between influenza and non-influenza cases of severe acute respiratory infections: A pilot study. Health Sci. Rep. 1 (6), e47. doi:10.1002/hsr2.47
Bosch, A. A. T. M., Levin, E., van Houten, M. A., Hasrat, R., Kalkman, G., Biesbroek, G., et al. (2016). Development of upper respiratory tract microbiota in infancy is affected by mode of delivery. EBioMedicine 9, 336–345. doi:10.1016/j.ebiom.2016.05.031
Bosch, A. A. T. M., de Steenhuijsen Piters, W. A. A., van Houten, M. A., Chu, M. L. J. N., Biesbroek, G., Kool, J., et al. (2017). Maturation of the infant respiratory microbiota, environmental drivers, and health consequences A prospective cohort study. Am. J. Respir. Crit. Care Med. 196, 1582–1590. doi:10.1164/rccm.201703-0554OC
Brook, I. (2001). Ocular infections due to anaerobic bacteria. Int. Ophthalmol. 24 (5), 269–277. doi:10.1023/A:1025431008020
Charlson, E. S., Chen, J., Custers-Allen, R., Bittinger, K., Li, H., Sinha, R., et al. (2010). Disordered microbial communities in the upper respiratory tract of cigarette smokers. PLoS ONE 5 (12), 15216. doi:10.1371/journal.pone.0015216
Charlson, E. S., Bittinger, K., Haas, A. R., Fitzgerald, A. S., Frank, I., Yadav, A., et al. (2011). Topographical continuity of bacterial populations in the healthy human respiratory tract. Am. J. Respir. Crit. Care Med. 184 (8), 957–963. doi:10.1164/rccm.201104-0655OC
Clausen, T. M., Sandoval, D. R., Spliid, C. B., Pihl, J., Perrett, H. R., Painter, C. D., et al. (2020). SARS-CoV-2 infection depends on cellular heparan sulfate and ACE2. Cell 183 (4), 1043–1057. doi:10.1016/J.CELL.2020.09.033
Cui, L., Morris, A., Huang, L., Beck, J. M., Iii, H. L. T., von Mutius, E., et al. (2014). The microbiome and the lung. Ann. Am. Thorac. Soc. 11, 227–232. doi:10.1513/AnnalsATS.201402-052PL
Cyprian, F., Sohail, M. U., Abdelhafez, I., Salman, S., Attique, Z., Kamareddine, L., et al. (2021). SARS-CoV-2 and immune-microbiome interactions: Lessons from respiratory viral infections. Int. J. Infect. Dis. 105, 540–550. doi:10.1016/j.ijid.2021.02.071
de Wit, E., van Doremalen, N., Falzarano, D., and Munster, V. J. (2016). SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 14 (8), 543–634. doi:10.1038/nrmicro.2016.81
Dickson, R. P., Erb-Downward, J. R., and Huffnagle, G. B. (2014). The role of the bacterial microbiome in lung disease. Expert Rev. Respir. Med. 7 (3), 245–257. doi:10.1586/ERS.13.24
Dutartre, H., Miller, W. A., Van, A., Kuyl, D., Khrustalev, V. V., Victorovich Khrustalev, V., et al. (2020). Translation-associated mutational U-pressure in the first ORF of SARS-CoV-2 and other coronaviruses. Front Microbiol. 11, 559165. doi:10.3389/fmicb.2020.559165
Edouard, S., Million, M., Bachar, D., Dubourg, G., Michelle, C., Ninove, L., et al. (2018). The nasopharyngeal microbiota in patients with viral respiratory tract infections is enriched in bacterial pathogens. Eur. J. Clin. Microbiol. Infect. Dis. 37 (9), 1725–1733. doi:10.1007/S10096-018-3305-8
Fan, J., Li, X., Gao, Y., Zhou, J., Wang, S., Huang, B., et al. (2020). The lung tissue microbiota features of 20 deceased patients with COVID-19. J. Clean. Prod. 81, e64–e67. doi:10.1016/J.JINF.2020.06.047
Feehan, A. K., Rose, R., Nolan, D. J., Spitz, A. M., Graubics, K., Colwell, R. R., et al. (2021). Nasopharyngeal microbiome community composition and structure is associated with severity of COVID-19 disease and breathing treatment. Appl. Microbiol. 1 (2), 177–188. doi:10.3390/applmicrobiol1020014
Golda, A., Malek, N., Dudek, B., Zeglen, S., Wojarski, J., Ochman, M., et al. (2011). Infection with human coronavirus NL63 enhances streptococcal adherence to epithelial cells. J. General Virol. 92 (6), 1358–1368. doi:10.1099/vir.0.028381-0
Goldstein, J. D., Godleski, J. J., Balikian, J. P., and Herman, P. G. (1982). Pathologic patternsof Serratia marcescens pneumonia. Human Pathol. 13 (5), 479–484.
Gorbalenya, A. E., Baker, S. C., Baric, R. S., de Groot, R. J., Drosten, C., Gulyaeva, A. A., et al. (2020). The species severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 5 (4), 536–544. doi:10.1038/s41564-020-0695-z
Gupta, R. K., George, R., and Nguyen-Van-Tam, J. S. (2008). Bacterial pneumonia and pandemic influenza planning. Emerg. Infect. Dis. 14 (8), 1187–1192. doi:10.3201/eid1408.070751
Gupta, A., Karyakarte, R., Joshi, S., Das, R., Jani, K., Shouche, Y., et al. (2022). Nasopharyngeal microbiome reveals the prevalence of opportunistic pathogens in SARS-CoV-2 infected individuals and their association with host types. Microbes Infect. 24 (1), 104880. doi:10.1016/J.MICINF.2021.104880
Guruprasad, L. (2021). Human SARS CoV-2 spike protein mutations. Proteins Struct. Funct. Bioinforma. 89 (5), 569–576. doi:10.1002/PROT.26042
Haiminen, N., Utro, F., Seabolt, E., and Parida, L. (2021). Functional profiling of COVID-19 respiratory tract microbiomes. Sci. Rep. 11 (1), 6433. doi:10.1038/s41598-021-85750-0
Hashimoto, T., Perlot, T., Rehman, A., Trichereau, J., Ishiguro, H., Paolino, M., et al. (2012). ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 487 (7408), 477–481. doi:10.1038/nature11228
Hikmet, F., Méar, L., Edvinsson, Å., Micke, P., Uhlén, M., and Lindskog, C. (2020). The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 16 (7), e9610. doi:10.15252/msb.20209610
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Krüger, N., Herrler, T., Erichsen, S., et al. (2020). SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181 (2), 271–280.e8. doi:10.1016/J.CELL.2020.02.052
Hu, B., Guo, H., Zhou, P., and Shi, Z.-L. (2020). Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 19, 141–154. doi:10.1038/s41579-020-00459-7
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395 (10223), 497–506. doi:10.1016/S0140-6736(20)30183-5
Huang, Y., Xie, J., Guo, Y., Sun, W., He, Y., Liu, K., et al. (2021). Sars-cov-2: Origin, intermediate host and allergenicity features and hypotheses. Healthc. Switz. 9 (9), 1132. doi:10.3390/healthcare9091132
Hyde, E. R., Andrade, F., Vaksman, Z., Parthasarathy, K., Jiang, H., Parthasarathy, D. K., et al. (2014). Metagenomic analysis of nitrate-reducing bacteria in the oral cavity: Implications for nitric oxide homeostasis. PLoS ONE 9 (3), 88645. doi:10.1371/journal.pone.0088645
Ibironke, O., McGuinness, L. R., Lu, S. E., Wang, Y., Hussain, S., Weisel, C. P., et al. (2020). Species-level evaluation of the human respiratory microbiome. GigaScience 9 (4), giaa038. doi:10.1093/GIGASCIENCE/GIAA038
Kan, B., Wang, M., Jing, H., Xu, H., Jiang, X., Yan, M., et al. (2005). Molecular evolution analysis and geographic investigation of severe acute respiratory syndrome coronavirus-like virus in palm civets at an animal market and on farms. J. Virol. 79 (18), 11892–11900. doi:10.1128/jvi.79.18.11892-11900.2005
Khan, A. A., and Khan, Z. (2020). COVID-2019-associated overexpressed Prevotella proteins mediated host-pathogen interactions and their role in coronavirus outbreak. Bioinformatics 36 (13), 4065–4069. doi:10.1093/bioinformatics/btaa285
Khan, R., Petersen, F. C., and Shekhar, S. (2019). Commensal bacteria: An emerging player in defense against respiratory pathogens. Front. Immunol. 10, 1203. doi:10.3389/fimmu.2019.01203
Kim, H. J., Jo, A., Jeon, Y. J., An, S., Lee, K.-M., Yoon, S. S., et al. (2019). Nasal commensal Staphylococcus epidermidis enhances interferon-λ-dependent immunity against influenza virus. Microbiome 7 (1), 80. doi:10.1186/s40168-019-0691-9
Kirtipal, N., Bharadwaj, S., and Kang, S. G. (2020). From SARS to SARS-CoV-2, insights on structure, pathogenicity and immunity aspects of pandemic human coronaviruses. Infect. Genet. Evol. 85, 104502. doi:10.1016/J.MEEGID.2020.104502
Kolhe, R., Shri Sahajpal, N., Vyavahare, S., Dhanani, A. S., Adusumilli, S., Ananth, S., et al. (2021). Alteration in nasopharyngeal microbiota profile in aged patients with COVID-19. Diagn. (Basel) 11, 1622. doi:10.3390/diagnostics11091622
Larsen, J. M., Steen-Jensen, D. B., Laursen, J. M., Søndergaard, J. N., Musavian, H. S., Butt, T. M., et al. (2012). Divergent pro-inflammatory profile of human dendritic cells in response to commensal and pathogenic bacteria associated with the airway microbiota. PLoS ONE 7 (2), e31976. doi:10.1371/journal.pone.0031976
Lau, S. K., Woo, P. C., Li, K. S., Huang, Y., Tsoi, H.-W., Wong, B. H., et al. (2005). Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. U. S. A. 102(39), 14040–5. doi:10.1073/pnas.0506735102
Li, Y., and Tang, X. X. (2021). Abnormal airway mucus secretion induced by virus infection. Front Immunol. 12, 701443. doi:10.3389/fimmu.2021.701443
Li, X., Geng, M., Peng, Y., Meng, L., and Lu, S. (2020). Molecular immune pathogenesis and diagnosis of COVID-19. J. Pharm. Anal. 10 (2), 102–108. doi:10.1016/J.JPHA.2020.03.001
Liu, D., Yumoto, H., Hirota, K., Murakami, K., Takahashi, K., Hirao, K., et al. (2008). Histone-like DNA binding protein of Streptococcus intermedius induces the expression of pro-inflammatory cytokines in human monocytes via activation of ERK1/2 and JNK pathways. Cell. Microbiol. 10 (1), 262–276. doi:10.1111/j.1462-5822.2007.01040.x
Liu, P., Jiangid, J.-Z., Wanid, X.-F., Hua, Y., Liid, L., Zhou, J., et al. (2020). Are pangolins the intermediate host of the 2019 novel coronavirus (SARS-CoV-2)? PLoS Pathog. 16, e1008421. doi:10.1371/journal.ppat.1008421
Liu, J., Liu, S., Zhang, Z., Lee, X., Wu, W., Huang, Z., et al. (2021). Association between the nasopharyngeal microbiome and metabolome in patients with COVID-19. Synthetic Syst. Biotechnol. 6 (3), 135–143. doi:10.1016/J.SYNBIO.2021.06.002
Lu, R., Zhao, X., Li, J., Niu, P., Yang, B., Wu, H., et al. (2020). Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 395 (10224), 565–574. doi:10.1016/S0140-6736(20)30251-8
Ludwig, S., and Zarbock, A. (2020). Coronaviruses and SARS-CoV-2: A brief overview. Anesth. Analg. 131 (1), 93–96. doi:10.1213/ANE.0000000000004845
Ma, S., Zhang, F., Zhou, F., Li, H., Ge, W., Gan, R., et al. (2021). Metagenomic analysis reveals oropharyngeal microbiota alterations in patients with COVID-19. Signal Transduct. Target. Ther. 6 (1), 191. doi:10.1038/s41392-021-00614-3
Macknight, G., Smith, L., Lamza, K., Mckenzie, H., Batt, L., Kelly, D., et al. (1997). Protection against oral and gastrointestinal diseases: Importance of dietary nitrate intake, oral nitrate reduction and enterosalivary nitrate circulation. Biochem. Physiol. 118 (4), 939–948. doi:10.1016/s0300-9629(97)00023-6
Martino, C., Kellman, B. P., Sandoval, D. R., Mandel Clausen, T., Marotz, C. A., Song, J., et al. (2020). Bacterial modification of the host glycosaminoglycan heparan sulfate modulates SARS-CoV-2 infectivity. BioRxiv. [Preprint]. doi:10.1101/2020.08.17.238444
Merenstein, C., Liang, G., Whiteside, S. A., Cobián-Güemes, A. G., Merlino, M. S., Taylor, L. J., et al. (2021). Signatures of COVID-19 severity and immune response in the respiratory tract microbiome. MBio 12 (4), e0177721. doi:10.1128/mBio.01777-21
Miyazaki, T. (2017). Protective effects of lactic acid bacteria on influenza A virus infection. AIMS Allergy Immunol. 1 (3), 138–142. doi:10.3934/Allergy.2017.3.138
Nardelli, C., Gentile, I., Setaro, M., di Domenico, C., Pinchera, B., Buonomo, A. R., et al. (2021). Nasopharyngeal microbiome signature in COVID-19 positive patients: Can we definitively get a role to fusobacterium periodonticum? Front. Cell. Infect. Microbiol. 11, 625581. doi:10.3389/fcimb.2021.625581
Olszak, T., An, D., Zeissig, S., Vera, M. P., Richter, J., Franke, A., et al. (2012). Microbial exposure during early life has persistent effects on natural killer T cell function. Science 336 (6080), 489–493. doi:10.1126/science.1219328
Payne, S. (2017). Family Coronaviridae. Viruses 2017, 149–158. doi:10.1016/b978-0-12-803109-4.00017-9
Ren, L., Wang, Y., Zhong, J., Li, X., Xiao, Y., Li, J., et al. (2021). Dynamics of the upper respiratory tract microbiota and its association with mortality in COVID-19. Am. J. Respir. Crit. Care Med. 204, 1379–1390. doi:10.1164/rccm.202103-0814OC
Rhoades, N. S., Pinski, A. N., Monsibais, A. N., Jankeel, A., Doratt, B. M., Cinco, I. R., et al. (2021). Acute SARS-CoV-2 infection is associated with an increased abundance of bacterial pathogens, including Pseudomonas aeruginosa in the nose. Cell Rep. 36 (9), 109637. doi:10.1016/J.CELREP.2021.109637
Ricciardolo, F. L. M., Sterk, P. J., Gaston, B., and Folkerts, G. (2004). Nitric oxide in health and disease of the respiratory system. Physiol. Rev. 84 (3), 731–765. doi:10.1152/physrev.00034.2003
Robinson, E., and Furlow, P. (2007). Anatomy of the respiratory system. Equine Respir. Med. Surg. 1, 3–17. doi:10.1016/B978-0-7020-2759-8.50006-4
Rosier, B. T., Buetas, E., Moya-Gonzalvez, E. M., Artacho, A., and Mira, A. (2020). Nitrate as a potential prebiotic for the oral microbiome. Sci. Rep. 10, 12895. doi:10.1038/s41598-020-69931-x
Rueca, M., Fontana, A., Bartolini, B., Piselli, P., Mazzarelli, A., Copetti, M., et al. (2021). Investigation of nasal/oropharyngeal microbial community of COVID-19 patients by 16S rDNA sequencing. Public Health 18, 2174. doi:10.3390/ijerph18042174
Sabir, J. S. M., Lam, T. T.-Y., Ahmed, M. M. M., Li, L., Shen, Y., Abo-Aba, E. M. S., et al. (2016). Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science 351 (6268), 81–84. doi:10.1126/science.aac8608
Santagati, M., Scillato, M., Patanè, F., Aiello, C., and Stefani, S. (2012). Bacteriocin-producing oral streptococci and inhibition of respiratory pathogens. FEMS Immunol. Med. Microbiol. 65 (1), 23–31. doi:10.1111/j.1574-695X.2012.00928.x
Shi, Y.-L., He, M.-Z., Han, M.-Z., Gui, H.-Y., Wang, P., Yu, J.-L., et al. (2022). Characterization of altered oropharyngeal microbiota in hospitalized patients with mild SARS-CoV-2 infection. Front. Cell. Infect. Microbiol. 12. doi:10.3389/fcimb.2022.824578
Shilts, M. H., Rosas-Salazar, C., Strickland, B. A., Kimura, K. S., Asad, M., Sehanobish, E., et al. (2022). Severe COVID-19 is associated with an altered upper respiratory tract microbiome. Front. Cell. Infect. Microbiol. 11, 781968. doi:10.3389/fcimb.2021.781968
Shukla, S. D., Budden, K. F., Neal, R., and Hansbro, P. M. (2017). Microbiome effects on immunity, health and disease in the lung. Clin. Transl. Immunol. 6 (3), e133. doi:10.1038/cti.2017.6
Smith, N., Goncalves, P., Charbit, B., Grzelak, L., Beretta, M., Planchais, C., et al. (2021). Distinct systemic and mucosal immune responses during acute SARS-CoV-2 infection. Nat. Immunol. 22 (11), 1428–1439. doi:10.1038/s41590-021-01028-7
Soltani, S., Hammami, R., Cotter, P. D., Rebuffat, S., ben Said, L., ene Gaudreau, H., et al. (2021). Bacteriocins as a new generation of antimicrobials: Toxicity aspects and regulations. FEMS Microbiol. Rev. 039 (1), 1–24. doi:10.1093/femsre/fuaa039
Stadio, A. D., Costantini, C., Renga, G., Pariano, M., Ricci, G., and Romani, L. (2020). The microbiota/host immune system interaction in the nose to protect from COVID-19. Life 10, 345. doi:10.3390/life10120345
Stubbendieck, R. M., May, D. S., Chevrette, M. G., Temkin, M. I., Wendt-Pienkowski, E., Cagnazzo, J., et al. (2019). Competition among nasal bacteria suggests a role for siderophore-mediated interactions in shaping the human nasal microbiota. Appl. Environ. Microbiol. 85 (10), e02406–e02418. doi:10.1128/AEM.02406-18
Tang, H.-J., Lai, C.-C., and Chao, C.-M. (2022). Changing epidemiology of respiratory tract infection during COVID-19 pandemic. Antibiotics 11 (3), 315. doi:10.3390/antibiotics11030315
Terrapon, N., Lombard, V., Gilbert, H. J., and Henrissat, B. (2015). Automatic prediction of polysaccharide utilization loci in Bacteroidetes species. Bioinformatics 31 (5), 647–655. doi:10.1093/bioinformatics/btu716
Tripathi, P., Tripathi, P., Kashyap, L., and Singh, V. (2007). The role of nitric oxide in inflammatory reactions. FEMS Immunol. Med. Microbiol. 51 (3), 443–452. doi:10.1111/j.1574-695X.2007.00329.x
Ventero, M. P., Cuadrat, R. R. C., Vidal, I., Andrade, B. G. N., Molina-Pardines, C., Haro-Moreno, J. M., et al. (2021). Nasopharyngeal microbial communities of patients infected with SARS-CoV-2 that developed COVID-19. Front. Microbiol. 12, 637430. doi:10.3389/fmicb.2021.637430
Wang, J., Li, F., Sun, R., Gao, X., Wei, H., Li, L.-J., et al. (2013). Bacterial colonization dampens influenza-mediated acute lung injury via induction of M2 alveolar macrophages. Nat. Commun. 4, 2106. doi:10.1038/ncomms3106
Wang, Y., Wang, Y., Luo, W., Huang, L., Xiao, J., Li, F., et al. (2020). A comprehensive investigation of the mrna and protein level of ace2, the putative receptor of sars-cov-2, in human tissues and blood cells. Int. J. Med. Sci. 17 (11), 1522–1531. doi:10.7150/ijms.46695
Wang, W., Tian, J.-H., Chen, X., Hu, R.-X., Lin, X.-D., Pei, Y.-Y., et al. (2022). Coronaviruses in wild animals sampled in and around Wuhan at the beginning of COVID-19 emergence. Virus Evol. 8, veac046. doi:10.1093/ve/veac046
Whelan, F. J., Verschoor, C. P., Stearns, J. C., Rossi, L., Luinstra, K., Loeb, M., et al. (2014). The loss of topography in the microbial communities of the upper respiratory tract in the elderly. Ann. Am. Thorac. Soc. 11 (4), 513–521. doi:10.1513/AnnalsATS.201310-351OC
Wink, D. A., Hines, H. B., Cheng, R. Y. S., Switzer, C. H., Flores-Santana, W., Vitek, M. P., et al. (2011). Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 89, 873–891. doi:10.1189/jlb.1010550
Xiong, D., Muema, C., Zhang, X., Pan, X., Xiong, J., Yang, H., et al. (2021). Enriched opportunistic pathogens revealed by metagenomic sequencing hint potential linkages between pharyngeal microbiota and COVID-19. Virol. Sin. 36, 924–933. doi:10.1007/s12250-021-00391-x
Yadav, R., Chaudhary, J. K., Jain, N., Chaudhary, P. K., Khanra, S., Dhamija, P., et al. (2021). Role of structural and non-structural proteins and therapeutic targets of SARS-CoV-2 for COVID-19. Cells 10, 821. doi:10.3390/cells10040821
Zhang, Y. Z., and Holmes, E. C. (2020). A genomic perspective on the origin and emergence of SARS-CoV-2. Cell 181 (2), 223–227. doi:10.1016/J.CELL.2020.03.035
Zhou, P., Yang, X.-L., Wang, X.-G., Hu, B., Zhang, L., Zhang, W., et al. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 270, 270. 273. doi:10.1038/s41586-020-2012-7
Ziebuhr, J., and Siddell, S. G. (1999). Processing of the human coronavirus 229E replicase polyproteins by the virus-encoded 3C-like proteinase: Identification of proteolytic products and cleavage sites common to pp1a and pp1ab. J. Virology 73 (1), 177–185. doi:10.1128/JVI.73.1.177-185.1999
Keywords: SARS-CoV-2, microbiome, respiratory tract, COVID-19, microbial diversity
Citation: Babišová K, Krumpolec P, Hadžega D, Sabaka P, Jackuliak P, Minárik G and Hýblová M (2023) Comparison of microbial diversity of respiratory tract between COVID-19 patients and healthy population. Acta Virol. 67:11664. doi: 10.3389/av.2023.11664
Received: 19 January 2023; Accepted: 09 May 2023;
Published: 22 June 2023.
Edited by:
Katarina Polcicovar, Slovak Academy of Sciences, SlovakiaCopyright © 2023 Babišová, Krumpolec, Hadžega, Sabaka, Jackuliak, Minárik and Hýblová. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Klaudia Babišová, a2xhdWRpYS5iYWJpc292YUBtZWRpcmV4Z3JvdXBhY2FkZW15LnNr