 Benjamin Dodd1,2
Benjamin Dodd1,2 Stephanie L. Moon1,2*
Stephanie L. Moon1,2*- 1Department of Human Genetics, University of Michigan, Ann Arbor, MI, United States
- 2Center for RNA Biomedicine, University of Michigan, Ann Arbor, MI, United States
Dystonia is a progressive neurological motor disease with few treatment options and no cure. This review synthesizes the results of recent studies that implicate protein kinase R in mediating the molecular mechanisms of dystonia pathogenesis. Mutations in the PKR gene EIF2AK2 and the PKR activator protein PACT are associated with early-onset generalized dystonia. Protein kinase R (PKR) is important for neuronal function. Genetic depletion or inhibition of PKR is associated with increased long-term potentiation and memory, while also causing neuronal hyper-excitability and seizures in mouse models. PKR also senses double stranded RNA within cells and activates the integrated stress response (ISR). The ISR is a conserved signaling pathway that hinges on controlled translational suppression to remodel gene expression during stress. When PKR is activated through binding double stranded RNA or the PKR activator protein PACT, PKR dimerizes, autophosphorylates, and phosphorylates the translation initiation factor eIF2. Translation suppression by p-eIF2 causes stress granule formation and the upregulation of stress-induced genes. The ISR is thought to drive cellular resilience during acute stress. However, chronic ISR activation is associated with neurological diseases, traumatic brain injury, and aging. Neurodevelopmental and neurodegenerative diseases are associated with mutations in other integrated stress response genes, suggesting a critical role for ISR regulation in neuronal health. A growing body of work suggests the ISR is also dysfunctional in dystonia. Future research investigating the molecular mechanisms of the ISR in dystonia will likely reveal therapeutic targets and treatment strategies for this currently incurable disease.
Introduction
Dystonia is a progressive neurological movement disorder that results in painful, uncontrolled muscle spasms with loss of motor function. The etiology of dystonia is complex and can be associated with genetic mutations or traumas such as traumatic brain injury. Human genetics data, studies on traumatic brain injury, and motor neurodegenerative conditions implicate the integrated stress response (ISR) as a critical pathway for neuronal dysfunction in dystonia pathogenesis. This article will focus on the evidence in the literature that suggests that dysregulation of the ISR signaling pathway contributes to the onset and progression of dystonia.
Alleles of the protein kinase R (PKR) gene EIF2AK2 and the PKR activator protein PACT [1] are associated with early-onset generalized dystonia. Missense variants in PKR are likely pathogenic because they were absent in gnomAD suggesting they are not found in the general population, they were observed in six unrelated families on three continents and are de novo, dominantly or recessively inherited, and they occur in the functional domain of PKR required for double stranded RNA binding [2, 3]. Early-onset generalized dystonia patients exhibited progressive muscle spasms and postural changes starting in the upper or lower extremities beginning in infancy or childhood [1]. Developmental delay and neurological symptoms including mild cognitive deficits, abnormal brain MRI, spasticity, and seizures were also observed in a subset of patients. Importantly, patients with this early-onset generalized dystonia experience seizures and neurological regression following a viral illness and/or fever [2, 3], suggesting that defects in the cellular stress response may underlie neuropathogenesis. In support of this idea, early-onset generalized dystonia patient-derived fibroblasts exhibit increased ISR signaling in long-term stress conditions [2]. Traumatic brain injury, a known trigger of dystonia, causes long-term, chronic activation of the ISR [4]. These and other observations implicate dysregulation of the ISR in the pathogenesis of dystonia.
The role of PKR in the integrated stress response
At the molecular level, PKR plays a critical role in activating the ISR by sensing stresses and globally suppressing protein biosynthesis in the cell. The ISR is a signaling pathway that modifies transcription and translation in response to intracellular signatures of stress. Acute activation of the ISR is thought to promote cell survival, while chronic ISR activation is proposed to drive cell death [5]. The ISR pathway progresses through four phases (Figure 1). First, stress-sensing protein kinases PKR, PERK (PKR-like endoplasmic reticulum kinase), GCN2 (general control non-derepressible 2), or HRI (heme-regulated inhibitor) are activated by molecular signatures that are proxies for underlying stresses including immune factors, proteostasis and endoplasmic reticulum defects, toxic metalloids, and pathogens. Protein kinase R binds double-stranded RNA (dsRNA) [6–9] and can be activated by protein-protein interactions (e.g., with PACT) during other stress conditions (such as endoplasmic reticulum stress) to initiate the ISR. Double-stranded RNA can accumulate within cells during viral infection and upon expression of endogenous genes that form complementary transcripts (e.g., repetitive nuclear sequences and endogenous retroviruses). These kinases dimerize, autophosphorylate, and phosphorylate the major translation initiation factor eIF2ɑ to inhibit translation initiation. Stress granules form from non-translating mRNAs and the first wave of stress-induced genes (such as ATF4) are selectively translated through upstream open reading frame (uORF) and/or non-canonical initiation mechanisms. Next, newly translated stress-induced transcription factors (e.g., ATF4) stimulate stress-induced gene transcription of uORF-mediated genes such as GADD34. Paradoxically, transcriptional targets of ATF4 include both adaptive genes that restore homeostasis and apoptotic genes that drive cell death [10]. Finally, the stress-induced gene GADD34 interacts with the serine/threonine protein phosphatase 1 PP1 to dephosphorylate p-eIF2ɑ and de-repress translation. Translation re-initiation corresponds with stress granule disassembly or clearance and amplifies stress-induced gene expression. Thus, PKR plays a critical role in activating the ISR pathway, which mediates cellular resilience to stress through reprogramming translation.
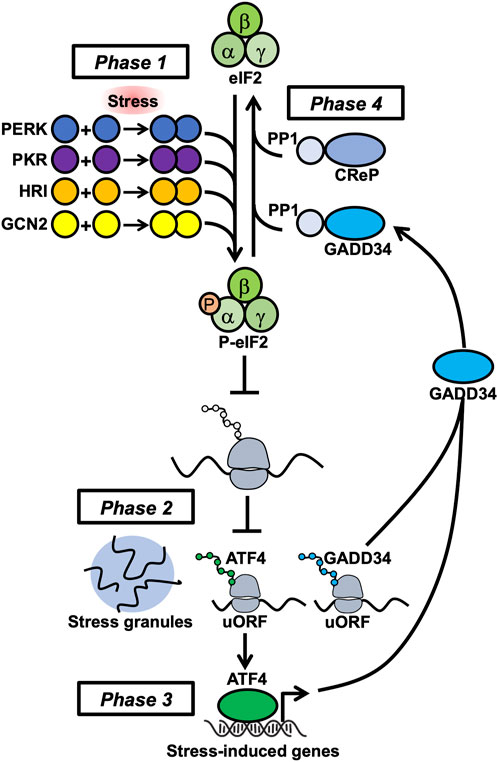
FIGURE 1. The four phases of the integrated stress response center on regulation of eIF2 phosphorylation.
The role of PKR in neurological function and disease
A growing body of work suggests that PKR plays important roles in neuronal function in the brain [2, 11–13]. Neuronal synapses harbor a unique proteome that drives neuronal excitability and synaptic plasticity to enable learning and memory [14, 15]. PKR is an important regulator of neuronal excitability. Additionally, PKR emerged evolutionarily in vertebrates as nervous systems gained complexity [16]. Deletion of the PKR gene or pharmacological inhibition of PKR enhances late long-term potentiation associated with increased learning and memory behaviors in murine models [12]. However, inhibition or depletion of PKR also results in neuronal network hyper-excitability in vitro and in vivo and seizures in a mouse model [12]. Forcing PKR dimerization in mouse hippocampal CA1 neurons inhibits late-long-term potentiation and is associated with impaired context-dependent memory [17]. Further, the target of PKR, eIF2ɑ, plays a role in memory consolidation that may occur through the regulation of local translation [18]. Phosphorylation of eIF2ɑ regulates late-long term potentiation and long-term memory through a mechanism that likely involves translation of specific mRNAs at the synapse. In line with these results, the small molecule inhibitor of the ISR that targets eIF2ɑ called ISRIB (integrated stress response inhibitor) similarly increases learning and memory in mice [19, 20]. Neurons have long distal axons and dendrites that can extend up to a meter from the cell body and rely heavily on local translation of RNAs that are trafficked to synapses for their function. Therefore, one possibility is that defects in neuronal function that contribute to dystonia pathogenesis could result in part from aberrant gene expression due to PKR dysfunction.
In addition to its role in neuronal function, activated PKR is observed in numerous neurodegenerative disease contexts. Brain tissue from patients with Creutzfeldt-Jakob’s, Parkinson’s, Huntington’s, Amyotrophic lateral sclerosis, and Alzheimer’s diseases exhibit markers of increased PKR activation [21–24]. Individuals with Down Syndrome have a dramatically increased risk of developing Alzheimer’s disease, and tissues from individuals with Down Syndrome have increased PKR activation [13]. Importantly, chemical or genetic suppression of PKR activity or the ISR rescued memory and synaptic plasticity phenotypes in a mouse model of Down Syndrome [13]. In addition to early-onset generalized dystonia, heterozygous missense mutations in the PKR gene are also associated with LEUDEN Syndrome, which is characterized by leukoencephalopathy, developmental delay, and neurologic decompensation [11]. Similarly to EIF2AK2 early-onset generalized dystonia, neurological regression was also observed in LEUDEN Syndrome patients following physiological stressors including viral infections and febrile illness that could cause PKR activation [11]. Beyond PKR signaling, chronic and/or severe ISR activation is associated with neurodegeneration [18], and mutations in ISR genes are associated with rare genetic developmental disorders [25]. Together, these studies suggest that precise regulation of PKR is important for neurological health.
Protein kinase R in dystonia
Protein kinase R dysfunction may cause early-onset dystonia pathogenesis. Human genetics data suggest four EIF2AK2 variants, Pro31Arg, Asn32Thr, Gly130Arg, and Gly138Ala are causative of early-onset generalized dystonia (Figure 2) [2, 3]. These EIF2AK2 variants were heterozygous or homozygous. Pedigree analysis of affected patients demonstrated de novo, dominant, or recessive inheritance of EIF2AK2 mutations associated with early-onset generalized dystonia. The Gly130Arg (c.388G>A) mutation was observed in nine patients from three unrelated families and was heterozygous or occurred de novo. The Gly138Ala (c. 413G>C) mutation was also heterozygous in a fourth affected family, suggesting these mutations are autosomal dominant. The Asn32Thr (c.95A>C) mutation was observed in a fifth affected family but was homozygous in the affected patient. The Pro31Arg (c.92C>G) mutation was observed in one heterozygous patient [3]. All four missense variants occur in the double-stranded RNA binding motifs of the PKR protein, which is critical for PKR function. Further, mutations in the PKR activator protein PACT have also been associated with a heritable dystonia [26–29].

FIGURE 2. Protein kinase R contains two double-stranded RNA binding motifs (DSRM) and a kinase domain. The four mutations in PKR associated with early-onset generalized dystonia are indicated in red.
Intriguingly, patients with EIF2AK2 early-onset generalized dystonia experience seizures and neurologic regression following viral and/or febrile illness that could drive the onset or progression of dystonia [2, 3]. Because PKR is a stress-sensing kinase that initiates the ISR, these results suggest physiological stresses in the context of PKR dysfunction might trigger dystonia. Beyond PKR in dystonia, mutations in the other stress-sensing kinases of the ISR pathway, GCN2, HRI, and PERK, are associated with neurological diseases (reviewed in [25]). Additionally, mutations in other ISR pathway genes including EIF2B genes are associated with progressive white matter loss that occurs with physiological stresses including febrile illness [18]. Rare mutations in ATF4, a key transcription factor induced by the integrated stress response pathway, are associated with sporadic cervical dystonia [30]. Together, the results of these studies suggest that dysregulation of PKR contributes to dystonia pathogenesis through altered ISR pathway activation.
Limited data in the literature suggest that EIF2AK2 mutations associated with early-onset generalized dystonia can hyperactivate PKR and upregulate the ISR [2]. Phosphorylation of PKR was four-to six-fold upregulated upon poly(I:C) stress (a double-stranded RNA mimic) in fibroblasts from early-onset generalized dystonia patients with either Asn32Thr or Gly130Arg mutations compared to unaffected controls. In line with this observation, these patient-derived fibroblasts exhibited an approximately three-fold increase in p-eIF2ɑ compared to unaffected controls upon poly(I:C) stress [2]. The implications of this study are that dystonia patient cells are hyper-sensitive to stress, which could lead to a persistent or irreversible ISR.
In addition to human genetics findings, several lines of evidence suggest that perturbation of the ISR pathway plays a key role in dystonia pathogenesis. Dysregulation of the stress response has been observed in models of other heritable dystonias including those caused by PACT, TOR1A, THAP1, and SGCE mutations [27, 30–32]. The results of in vitro assays using purified PKR suggest dystonia-associated PACT mutations cause PKR hyperphosphorylation [27]. Further, analysis of lymphoblasts from one DYT16 patient suggests persistent eIF2ɑ phosphorylation after endoplasmic reticulum stress could contribute to dystonia pathogenesis [27]. Intriguingly, a genome-wide siRNA screen to rescue subcellular localization defects of a mutant TOR1A associated with DYT1 in tissue culture cells revealed eIF2ɑ signaling was the top enriched pathway among hits [30]. Depletion of PKR or other eIF2ɑ kinases worsened the mutant TOR1A localization defect, while blocking p-eIF2ɑ dephosphorylation pharmacologically with Salubrinal rescued TOR1A subcellular localization [30]. Strikingly, Salubrinal treatment improved neonatal survival of a mouse model of DYT1 and rescued long-term synaptic depression in corticostriatal neurons in brain slices [30]. The eIF2ɑ signaling pathway was significantly dysregulated in brain tissues of Thap1± mice in a model of DYT6 and a derivative of Salubrinal rescued long-term synaptic depression in brain slices [31]. Additionally, the PKR gene Eif2ak2 and the stress-induced gene Fos were significantly upregulated in cerebellar tissue from a mouse model of SGCE myoclonus dystonia [32]. Together, these data suggest that dysregulation of the ISR could contribute to progressive neurological disorders including dystonia.
Discussion
An emerging area of research suggests a critical role for protein kinase R and the integrated stress response in dystonia pathogenesis. Human genetics data suggest mutations in the genes encoding PKR or the PKR activator protein PACT associated with early-onset generalized dystonia increase PKR function by hyperactivation of the ISR. Two molecular mechanisms could underlie this observation. First, PKR mutations could increase PKR activity, sensitizing the cell to stress and hyper-activating the ISR. Such a mechanism could occur if the mutations increase the binding affinity of PKR to RNA, increase the affinity of PKR homodimerization, increase PKR kinase activity, or reduce the inactivation or degradation of the activated PKR dimer. Such a mechanism is likely considering the observations that PKR and eIF2ɑ are hyper-phosphorylated in poly(I:C)-stressed cells derived from early-onset dystonia patients. Yet, a second possibility is that mutations in PKR associated with dystonia decrease PKR function and cause an irreversible ISR. Such a mechanism may be less likely to occur because early-onset dystonia patient fibroblasts exhibited increased phospho-PKR levels upon poly(I:C) stress [2]. Because PKR plays additional roles in neuronal excitability, an important area of future research will be in elucidating the mechanisms by which neuronal function are impaired by PKR hyperactivation. These possible mechanisms must be interrogated experimentally to understand the mechanisms of generalized dystonia, elucidate diverse neurological phenotypes associated with PKR dysfunction, and pinpoint therapeutic intervention strategies for rational drug design. Not only will such research shed light on the molecular mechanisms of dystonia, but this area of work holds promise for revealing common aspects of neurodegenerative and neurodevelopmental disorders.
Author contributions
BD and SM wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The Dystonia Medical Research Foundation (DMRF-MCMD-2022-1 to SM) supported this work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Dystonia Medical Research Foundation for supporting this work (award to SM). Figure 2 was created using BioRender.com.
References
1. Patel, RC, and Sen, GC. PACT, a protein activator of the interferon-induced protein kinase, PKR. PKR EMBO J (1998) 17(15):4379–90. doi:10.1093/emboj/17.15.4379
2. Kuipers, DJS, Mandemakers, W, Lu, C-S, Olgiati, S, Breedveld, GJ, Fevga, C, et al. EIF2AK2 missense variants associated with early onset generalized dystonia. Ann Neurol (2021) 89(3):485–97. doi:10.1002/ana.25973
3. Waller, SE, Morales-Briceño, H, Williams, L, Mohammad, SS, Fellner, A, Kumar, KR, et al. Possible EIF2AK2-associated stress-related neurological decompensation with combined dystonia and striatal lesions. Mov Disord Clin Pract (2022) 9(2):240–4. doi:10.1002/mdc3.13384
4. Chou, A, Krukowski, K, Jopson, T, Zhu, PJ, Costa-Mattioli, M, Walter, P, et al. Inhibition of the integrated stress response reverses cognitive deficits after traumatic brain injury. Proc Natl Acad Sci U S A (2017) 114(31):E6420–6. doi:10.1073/pnas.1707661114
5. Pakos-Zebrucka, K, Koryga, I, Mnich, K, Ljujic, M, Samali, A, and Gorman, AM. The integrated stress response. EMBO Rep (2016) 17(10):1374–95. doi:10.15252/embr.201642195
6. Meurs, E, Chong, K, Galabru, J, Thomas, NS, Kerr, IM, Williams, BR, et al. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell (1990) 62(2):379–90. doi:10.1016/0092-8674(90)90374-n
7. Katze, MG, Wambach, M, Wong, ML, Garfinkel, M, Meurs, E, Chong, K, et al. Functional expression and RNA binding analysis of the interferon-induced, double-stranded RNA-activated, 68,000-Mr protein kinase in a cell-free system. Mol Cell Biol (1991) 11(11):5497–505. doi:10.1128/mcb.11.11.5497
8. Patel, RC, and Sen, GC. Identification of the double-stranded RNA-binding domain of the human interferon-inducible protein kinase. J Biol Chem (1992) 267(11):7671–6. doi:10.1016/s0021-9258(18)42567-7
9. Green, SR, Manche, L, and Mathews, MB. Two functionally distinct RNA-binding motifs in the regulatory domain of the protein kinase DAI. Mol Cell Biol (1995) 15(1):358–64. doi:10.1128/mcb.15.1.358
10. Wortel, IMN, van der Meer, LT, Kilberg, MS, and van Leeuwen, FN. Surviving stress: modulation of ATF4-mediated stress responses in normal and malignant cells. Trends Endocrinol Metab (2017) 28(11):794–806. doi:10.1016/j.tem.2017.07.003
11. Mao, D, Reuter, CM, Ruzhnikov, MRZ, Beck, AE, Farrow, EG, Emrick, LT, et al. De novo EIF2AK1 and EIF2AK2 variants are associated with developmental delay, leukoencephalopathy, and neurologic decompensation. Am J Hum Genet (2020) 106(4):570–83. doi:10.1016/j.ajhg.2020.02.016
12. Zhu, PJ, Huang, W, Kalikulov, D, Yoo, JW, Placzek, AN, Stoica, L, et al. Suppression of PKR promotes network excitability and enhanced cognition by interferon-γ-mediated disinhibition. Cell (2011) 147(6):1384–96. doi:10.1016/j.cell.2011.11.029
13. Zhu, PJ, Khatiwada, S, Cui, Y, Reineke, LC, Dooling, SW, Kim, JJ, et al. Activation of the ISR mediates the behavioral and neurophysiological abnormalities in Down syndrome. Science (2019) 366(6467):843–9. doi:10.1126/science.aaw5185
14. Glock, C, Biever, A, Tushev, G, Bartnik, I, Assir, BN, Dieck, ST, et al. The mRNA translation landscape in the synaptic neuropil. bioRxiv (2020). Available from: https://www.biorxiv.org/content/10.1101/2020.06.09.141960v1 (Accessed June 10, 2020).
15. Kiebler, MA, and Bassell, GJ. Neuronal RNA granules: movers and makers. Neuron (2006) 51(6):685–90. doi:10.1016/j.neuron.2006.08.021
16. Taniuchi, S, Miyake, M, Tsugawa, K, Oyadomari, M, and Oyadomari, S. Integrated stress response of vertebrates is regulated by four eIF2α kinases. Sci Rep (2016) 6:32886. doi:10.1038/srep32886
17. Jiang, Z, Belforte, JE, Lu, Y, Yabe, Y, Pickel, J, Smith, CB, et al. eIF2alpha Phosphorylation-dependent translation in CA1 pyramidal cells impairs hippocampal memory consolidation without affecting general translation. J Neurosci (2010) 30(7):2582–94. doi:10.1523/JNEUROSCI.3971-09.2010
18. Moon, SL, Sonenberg, N, and Parker, R. Neuronal regulation of eIF2α function in health and neurological disorders. Trends Mol Med (2018) 24(6):575–89. doi:10.1016/j.molmed.2018.04.001
19. Krukowski, K, Nolan, A, Frias, ES, Boone, M, Ureta, G, Grue, K, et al. Small molecule cognitive enhancer reverses age-related memory decline in mice. Elife (2020) 9:e62048. doi:10.7554/eLife.62048
20. Sidrauski, C, Acosta-Alvear, D, Khoutorsky, A, Vedantham, P, Hearn, BR, Li, H, et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife (2013) 2:e00498. doi:10.7554/eLife.00498
21. Peel, AL, and Bredesen, DE. Activation of the cell stress kinase PKR in Alzheimer’s disease and human amyloid precursor protein transgenic mice. Neurobiol Dis (2003) 14(1):52–62. doi:10.1016/s0969-9961(03)00086-x
22. Bando, Y, Onuki, R, Katayama, T, Manabe, T, Kudo, T, Taira, K, et al. Double-strand RNA dependent protein kinase (PKR) is involved in the extrastriatal degeneration in Parkinson’s disease and Huntington’s disease. Neurochem Int (2005) 46(1):11–8. doi:10.1016/j.neuint.2004.07.005
23. Paquet, C, Bose, A, Polivka, M, Peoc’h, K, Brouland, JP, Keohane, C, et al. Neuronal phosphorylated RNA-dependent protein kinase in Creutzfeldt-Jakob disease. J Neuropathol Exp Neurol (2009) 68(2):190–8. doi:10.1097/NEN.0b013e318196cd7c
24. Zu, T, Guo, S, Bardhi, O, Ryskamp, DA, Li, J, Khoramian Tusi, S, et al. Metformin inhibits RAN translation through PKR pathway and mitigates disease in C9orf72 ALS/FTD mice. Proc Natl Acad Sci U S A (2020) 117(31):18591–9. doi:10.1073/pnas.2005748117
25. English, AM, Green, KM, and Moon, SL. A (dis)integrated stress response: genetic diseases of eIF2α regulators. Wiley Interdiscip Rev RNA (2021) 13:e1689. doi:10.1002/wrna.1689
26. Camargos, S, Scholz, S, Simón-Sánchez, J, Paisán-Ruiz, C, Lewis, P, Hernandez, D, et al. DYT16, a novel young-onset dystonia-parkinsonism disorder: identification of a segregating mutation in the stress-response protein PRKRA. Lancet Neurol (2008) 7(3):207–15. doi:10.1016/S1474-4422(08)70022-X
27. Burnett, SB, Vaughn, LS, Sharma, N, Kulkarni, R, and Patel, RC. Dystonia 16 (DYT16) mutations in PACT cause dysregulated PKR activation and eIF2α signaling leading to a compromised stress response. Neurobiol Dis (2020) 146:105135. doi:10.1016/j.nbd.2020.105135
28. Vaughn, LS, Bragg, DC, Sharma, N, Camargos, S, Cardoso, F, and Patel, RC. Altered activation of protein kinase PKR and enhanced apoptosis in dystonia cells carrying a mutation in PKR activator protein PACT. J Biol Chem (2015) 290(37):22543–57. doi:10.1074/jbc.M115.669408
29. Burnett, SB, Vaughn, LS, Strom, JM, Francois, A, and Patel, RC. A truncated PACT protein resulting from a frameshift mutation reported in movement disorder DYT16 triggers caspase activation and apoptosis. J Cell Biochem (2019) 120(11):19004–18. doi:10.1002/jcb.29223
30. Rittiner, JE, Caffall, ZF, Hernández-Martinez, R, Sanderson, SM, Pearson, JL, Tsukayama, KK, et al. Functional genomic analyses of mendelian and sporadic disease identify impaired eIF2α signaling as a generalizable mechanism for dystonia. Neuron (2016) 92(6):1238–51. doi:10.1016/j.neuron.2016.11.012
31. Zakirova, Z, Fanutza, T, Bonet, J, Readhead, B, Zhang, W, Yi, Z, et al. Mutations in THAP1/DYT6 reveal that diverse dystonia genes disrupt similar neuronal pathways and functions. PLoS Genet (2018) 14(1):e1007169. doi:10.1371/journal.pgen.1007169
Keywords: protein kinase R, integrated stress response, EIF2AK2, dystonia, translation regulation
Citation: Dodd B and Moon SL (2023) The role of protein kinase R in dystonia. Dystonia 2:11718. doi: 10.3389/dyst.2023.11718
Received: 21 June 2023; Accepted: 14 September 2023;
Published: 22 September 2023.
Edited by:
Jan K. Teller, Scientific Advisors International, PolandCopyright © 2023 Dodd and Moon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephanie L. Moon, c21zbG1vb25AdW1pY2guZWR1