
Mechanisms of GNAL linked dystonia
 Mark S. Moehle
Mark S. Moehle- Department of Pharmacology and Therapeutics and Center for Translational Research in Neurodegeneration, University of Florida College of Medicine, Gainesville, FL, United States
Mutations in the GNAL gene, encoding Gαolf, are causative for an adult-onset, isolated dystonia that may provide unique insights into the etiology of adult-onset idiopathic dystonia. Gαolf is an alpha subunit of heterotrimeric G protein that replaces Gαs in the striatum and has unique expression patterns outside of the striatum. Gαolf additionally has defined molecular functions in GPCR signaling. These defined molecular pathways and expression pathways point to defined circuit deficits underlying the causes of this adult-onset dystonia that may provide additional insights into broader idiopathic dystonia. Here, we will review the available evidence for normal Gαolf function, and how this is corrupted by GNAL mutations to cause dystonia. Thes include the molecular signaling and expression profiles of Gαolf and the other G proteins, β2γ7, complexedwith it., Additionally, we will discuss the circuits that Gαolf influences, and how GNAL mutations may reorganize these circuits to cause dystonia.
Introduction
In 2013, the first mutations in GNAL were reported through whole exome sequencing in 2 families with dystonia which revealed a nonsense mutation in one family and a missense mutation in the other [1]. Further screening in 39 other families with dystonia revealed another 6 autosomal dominant mutations in GNAL including nonsense, missense, frameshift, and deletions [1]. The frequency of mutations in these families was 19%, and suggested that mutations in GNAL may be a common cause of familial dystonia. Subsequent studies have revealed at least 30 more pathogenic variants in GNAL as well as rare autosomal recessive mutations that are linked to dystonia [1–8]. Additionally, recent evidence has also suggested that sporadic GNAL mutations are linked to dystonia, and that >50% of all GNAL dystonia cases are sporadic [8–10]. These genetic studies have suggested that GNAL linked dystonia is rarer than originally believed with a frequency as low as 0.5% of familial dystonia attributed to mutations in GNAL [3].
Clinically, patients with mutations in GNAL are largely indistinguishable from idiopathic dystonia [1–5, 10, 11]. Age of onset in patients is typically in adulthood with age range at diagnosis between 7 and 68 years (average ages 31–42 years) [1, 8, 11]. Also, GNAL dystonia patients present with an isolated dystonia that often starts focally, sometimes becomes segmental, and rarely becomes generalized [8]. The exception to this is the rare autosomal recessive mutations in GNAL which also present with intellectual disabilities [6]. This high overlap in clinical characteristics with idiopathic disease, as well as defined gene function of GNAL discussed below, may make this form of genetic dystonia an interesting model system to understand synapses, circuits, and cells that are disturbed in dystonia, and how these alterations can be manipulated for novel therapeutic targets. Here, we review the known mechanisms of GNAL mutations, the functions of its protein product Gαolf, and discuss how these mutations may lead to dystonia.
Molecular signaling and expression of Gαolf
GNAL encodes Gαolf which is an alpha subunit of a heterotrimeric G protein [3, 12]. Gαolf is part of the GαS family of alpha subunits [13]. Upon neurotransmitter binding to the G protein coupled receptor, Gαolf switches from a GDP bound state to GTP bound state, dissociates from the receptor, and the G βγ subunits. Active Gαolf then binds to adenylate cyclase type 5, activates this enzyme, and leads to the production of the second messenger cAMP which then acts on its many downstream targets including protein kinase A, exchange proteins activated by cAMP (EPAC proteins), and cyclic nucleotide gated ion channels (See Figure 1) [14]. In order to inactivate Gαolf, GTP must be hydrolyzed to GDP which can achieved by the slow intrinsic GTPase activity of Gαolf and can be accelerated by regulator of G signaling (RGS) proteins [13]. Normal signaling of Gαolf is critical to the function of the striatum as Gαolf is the determining factor in cAMP second messenger production in the striatum [15–18]. Gαolf has an expression pattern that points to a unique role for the above signaling in normal basal ganglia function. Gαolf replaces GαS in the predominant neuron types in the striatum, spiny projection neurons. Additionally, Gαolf is co-expressed with Gαs in cholinergic interneurons, and possibly other interneuron classes, in the striatum [8, 19, 20]. This indicates that Gαolf is the major signal transducing alpha subunit for pro-excitatory neurotransmitters and their G protein coupled receptors in the striatum with Gαolf coupling to the D1 dopamine receptor in direct pathway spiny projection neurons and the adenosine 2A receptor in indirect pathway spiny projection neurons [15]. While the signaling role for Gαolf in the striatum is better defined, the role for Gαolf outside of the striatum is not well understood. Gαolf is expressed in multiple brain nuclei. IHC studies indicate Gαolf expression in Purkinje Cells of the cerebellum and dopaminergic cells of the substantia nigra pars compacta [8]. RNAseq and proteomics studies have supported even further widespread expression including in multiple cortical regions, thalamus, and amygdala [21–23]. However, receptors to which Gαolf couples in these regions are not understood, and in many cases the cell type or types that express Gαolf in these regions are not well understood. Understanding the complete expression profile of Gαolf with cell type specificity will aid in understanding the mechanisms and networks that are corrupted by mutations in GNAL to lead to dystonia.
Figure 1
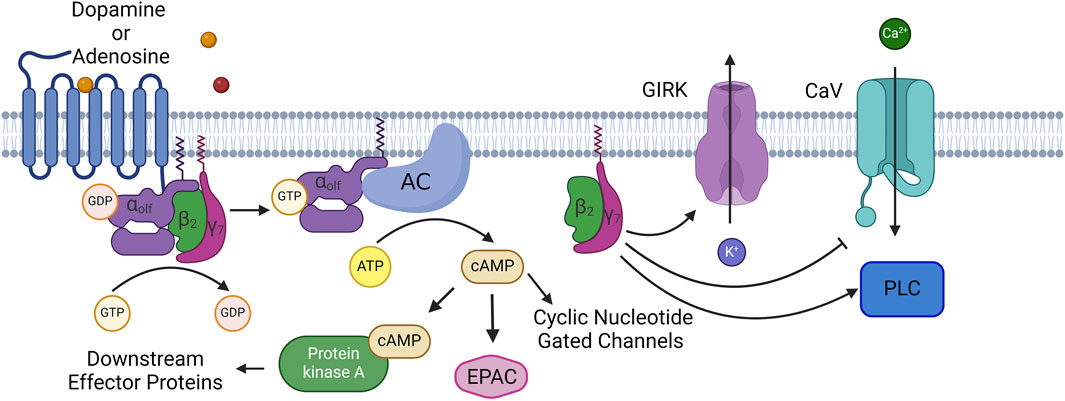
Figure 1. Normal Signaling Pathway of Gαolf: At rest Gαolf is complexed with βγ subunits (β2γ7 in spiny projection neurons) into an inactive heterotrimer. When neurotransmitter binds to its GPCR, Gαolf switches GDP for GTP, causing the heterotrimer to disassociate and its components are now active. Gαolf will bind to and activate adenylate cyclase 5, and leads to the production and accumulation of the second messenger cAMP. cAMP has a number of effector proteins including protein kinase A (PKA), exchange proteins activated by cAMP (EPAC), and cyclic nucleotide gated channels. β2γ7 also will modulate the activity of several enzymes and channels. These include G-protein gated inward-rectifying potassium channels (GIRK), voltage gated calcium channels, N, P, and Q type calcium channels, as well as phospholipase C. Created with BioRender.com.
Mechanisms of mutations in Gαolf
In the original paper describing the first mutations in GNAL, bioluminescence resonance energy transfer experiments gave the first evidence that disease associated mutations were loss of function mutations [1]. These experiments tagged the βγ subunits associated with Gαolf with the venus acceptor in this system and a downstream effector of βγ, GIRK, with the luciferase donor. Upon stimulation of the D1 dopamine receptor, mutations showed decreased association of βγ with GIRK, and suggested that mutations in GNAL represent loss of function mutations as they disrupt normal dopamine signaling through the D1 dopamine receptor [1]. However, the original description of the effects of GNAL mutations did not examine Gαolf dependent functions such as cAMP production.
More recently, however, the effects of GNAL mutations on Gαolf function have been examined in exquisite detail [12]. As discussed above, Gαolf is critical not only for the activation of adenylate cyclase and the production of cAMP, but also in formation of the heterotrimeric G protein complex, sequestering βγ from being active, and also termination of the signal through hydrolysis of GTP [12, 13]. These studies rigorously examined the effects of the known mutations at the time of the study on the full spectrum of molecular events that Gαolf is associated with. These studies revealed that, with respect to normal, well-regulated dopamine signaling, known mutations in GNAL are loss of function, but that the effects of the mutations on Gαolf function are significantly more nuanced than previously appreciated.
Each of the mutations had distinct effects on stability of Gαolf, formation of the trimer, G protein activation, signal termination, cAMP production, and basal signaling which led to each mutation having a distinct functional phenotype [12]. Interestingly, some of the mutations led to increased basal or dopamine induced cAMP production which would more traditionally appear as gain of function mutations. However, despite this increased basal or induced cAMP production, these mutations still represent a loss of function with respect to normal response to dopamine release [12]. This challenges the simple assignment of gain or loss of function to GNAL linked dystonia, and highlights the complicated molecular mechanisms behind mutations in GNAL. Furthermore, when these differential mutation effects on Gαolf are compared to dystonia severity or symptom clusters, the mutation effects do not significantly associate with any phenotype [12]. This suggests that although diverse in their mechanism, mutations in GNAL lead to an isolated dystonia phenotype through diverging from a narrow range of normal Gαolf activity.
Signaling partners of Gαolf also are associated with dystonia
Giving further validity to the critical nature of normal levels of Gαolf signaling are further human genetic studies that point to proteins both upstream and downstream of Gαolf as causative for dystonia. Mutations in five genes (GCH1, TH, PTS, SPR, and QDPR) that lead to disruption of the synthesis of dopamine are causative for dystonia [24–28]. Also upstream of Gαolf, mutations in the D1 dopamine receptor (DRD1) are linked to infantile parkinsonism-dystonia and tardive-like dystonia [29, 30]. Downstream of Gαolf, there are dystonia associated mutations in ADCY5, encoding adenylate cyclase 5, the enzyme responsible for the second messenger cAMP [31]. Further downstream enzymes, including PDE2A and PDE10A which are phosphodiesterase’s responsible for metabolizing cAMP to AMP are causative for a neurological syndrome that includes dystonia as a symptom [32, 33]. Also, mutations in DARPP-32, a signal transducing protein of the cAMP effector protein kinase A are dystonia linked [34]. These mutations upstream and downstream of Gαolf show that disrupting normal dopamine signaling can cause dystonia. However, there is one key difference between GNAL mutations and mutations for the other proteins in this pathway, Other than rare bi-allelic autosomal recessive mutations in GNAL [6], these autosomal dominant mutations in GNAL lead to an isolated dystonia which is usually adult onset. However, these other mutations in the Gαolf pathway often produce a neurological disorder that has dystonia as a symptom of a larger clinical syndrome, rather than in isolation, with juvenile age of onset. Why GNAL mutations are unique in this regard are not known, but point to a unique opportunity for utilizing GNAL to understand the pathophysiology of dystonia.
Gαolf dysfunction is associated with other movement disorders
While not genetically linked to other movement disorders, alterations in expression and activity of Gαolf are associated with Parkinson’s Disease and treatment induced dyskinesia [35]. This is most notable in levodopa induced dyskinesia, which is an adverse effect of the gold-standard of treatment for Parkinson’s disease. In levodopa induced dyskinesia, expression levels of Gαolf are associated with development and severity of dyskinesia in experimental models of this disorder, and not upstream receptors or neurotransmitters [35–38]. This points to an important role of Gαolf activity in the regulation of normal movement, and also a unique role of aberrant Gαolf activity in producing abnormal movement. However, how Gαolf activity can differentially alter striatal output and basal ganglia activity to produce different disease states is not known.
Possible βγ contributions to GNAL dystonia
Gα subunits in the GDP bound state sequester Gβγ, and prevent the Gβγ subunits from interacting with their effector proteins [13]. In the striatum, Gαolf couples to β2γ7 to form a form the functional heterotrimeric G protein [17, 18]. While the levels of expression of individual components of the heterotrimeric G protein are linked, decreased expression or removal of a single component the heterotrimeric G protein does not completely eliminate the other G protein components. For example, genetic removal of Gαolf or γ7 reduces the expression of the other, but does not eliminate it [12, 39]. Additionally, even in GNAL mutations that increase turnover of Gαolf or reduces stability of the trimer, there is still observable expression of β2γ7 [12]. This has led to all mutations in GNAL having observable unsequestered β2γ7 [12]. The consequences of this unsequestered and freely active β2γ7 are unknown. However, there are several possibilities. βγ subunits modulate the activity of several effector proteins including activation of phospholipase C, activation of GIRK channels, and inhibition of N, P, and Q type calcium channels [40–43]. Interestingly, one of the primary effectors of Gβγ, GIRK, are not expressed in striatal spiny projection neurons [44]. Unregulated modulation of these effectors by unsequestered β2γ7 could have profound effects on neuronal physiology and alter striatal activity. However, how unsequestered β2γ7, and possibly other βγ subunits in cells expressed outside the striatum, contribute to the development, maintenance, or expression of dystonia remains untested.
Models of GNAL linked dystonia
Gαolf expression and function were originally described in the olfactory epithelium where it is a key mediator of odorant receptor signaling. A global knockout of GNAL was made to study Gαolf in this context, and found that homozygous knockout of GNAL results in anosmia [19]. Also, due to feeding deficits, the majority of homozygous knockout mice die in early postnatal development. The original report of these mice showed that these homozygous knockout mice were paradoxically hyperactive, but the effects of GNAL loss on motor systems was confounded by the failure to thrive of global knockout mice that survived post weaning [19]. Heterozygous mice, however, are viable, have normal olfactory function, and are of comparable weight to wildtype littermate controls. These heterozygous mice do not display alterations in gross locomotor ability, but do have rotarod deficits that are progressive with age [45]. More recently, a heterozygous GNAL rat was developed, but displays similar motor deficits to the mouse with decreased spontaneous locomotion and rotarod deficits [46]. However, both the mouse and the rat GNAL model do not display overt dystonia like motor deficits in both visual observation based assays as well as EMG [45, 46].
Key findings from rodent models of dystonia
Despite the general lack of overt dystonic symptoms in the GNAL heterozygous mice and rats, these models have driven valuable understanding of how loss of Gαolf alters the central nervous system, and have given possible electrophysiological and biochemical endophenotypes of neuronal dysfunction to link to dystonic symptoms. First, the GNAL heterozygous mouse can have abnormal movements induced by administration of the non-selective muscarinic acetylcholine receptor agonist oxotremorine either through intraperitoneal injection or microinjection into the dorsal striatum but not the cerebellum [45]. GNAL heterozygous mice are more sensitive than littermate controls to administration of oxotremorine, regardless of route of administration, which points to an important role for cholinergic interneurons of the striatum in regulating GNAL linked dystonia.
Second, rodent models have pointed to electrophysiological alterations in basal ganglia physiology that may be important to the generation of dystonic motor phenotypes. Interestingly, GNAL heterozygosity did not alter intrinsic properties of striatal spiny projection neurons in the rat model of GNAL with no changes in resting membrane potential, membrane resistance, or rheobase current [46]. Additionally, no baseline differences in cortico-striatal evoked post-synaptic currents or paired pulse ratio was observed. However, there was significant impairment of cortico-striatal long-term depression that was partially ameliorated through application of D1 and D2 dopamine receptor agonists and fully ameliorated through Adenosine2A receptor agonists [46]. These electrophysiological findings have pointed to a potential unique role in altering activity dependent striatal activity, and altering striatal processing of cortical inputs.
Next, rodent models of GNAL have pointed to nuclei outside of basal ganglia as being altered by GNAL heterozygosity. As discussed above, Gαolf is expressed in Purkinje Cells of the cerebellum [20]. Using in vivo electrophysiology and optogenetics, recent evidence has shown that in both an asymptomatic state and oxotremorine induced abnormal movement state, cerebellar connectivity to cortical and thalamic nuclei is altered [47]. At baseline, GNAL heterozygous mice have decreased cerebello-thalamic plasticity induced by deep cerebellar nuclei stimulation [47]. Also, after abnormal movements were induced through oxotremorine administration, both cortical and thalamic inputs from the cerebellum were altered after deep cerebellar nuclei stimulation [47]. Interestingly, this alteration in cerebellar-thalamic activity may drive striatal dysfunction as well through decreased connectivity to fast spiking interneurons in GNAL heterozygous mice [47].
However, an important caveat to all of these key findings is the lack of overt dystonic symptoms in these models which prevents the unambiguous link between electrophysiological and biochemical endophenotypes to dystonic symptoms. Development of rodent models that harbor the patient derived mutations in GNAL or a Gnal floxed mouse that allows for the conditional removal of Gnal may prevent the post-natal lethality of the global Gnal knockout mouse, and allow for the development of dystonia like movements in mice. This will be critical for understanding of the neuronal changes induced by mutations in GNAL to cause dystonia.
Targeting Gαolf for therapeutic benefit
Due to levels and activity of Gαolf being associated with other movement disorders, directly targeting Gαolf with small molecules may not be the optimal strategy to target GNAL linked dystonia [36, 37]. Also, due to the diversity of mechanisms of GNAL mutations, directly targeting Gαolf may prove impractical as well [12]. Instead, targeting known modifiers of Gαolf function may provide greater efficacy. One possibility is the M4 muscarinic acetylcholine receptor [48, 49]. M4 directly opposes Gαolf and D1 dopamine receptor activity in direct pathway spiny projection neurons with M4 activation inhibiting adenylate cyclase and subsequent cAMP production [50]. Removing this inhibition of adenylate cyclase in direct pathway spiny projection neurons through M4 selective inhibitors may help to restore normal striatal output and basal ganglia activity to reduce dystonic symptoms [48, 49, 51]. Currently used small molecule therapeutics for dystonia may point to this treatment path as being promising. The non-selective muscarinic antagonist trihexyphenidyl remains a mainstay of treatment options for dystonia, but is not well tolerated by patients due to adverse effects [52, 53]. Selective M4 antagonists may provide efficacy in GNAL linked dystonia without the adverse effects of non-selective muscarinic antagonists. The first series of truly M4 selective antagonists have recently been developed [54], and the extent of their potential efficacy and liabilities has been highlighted in recent reviews [48, 49].
The defined signaling pathway of Gαolf also provides several additional possible therapeutic strategies for GNAL linked dystonia. Targeting modifiers of Gαolf which are downstream of Gαolf itself will likely be important, as targeting upstream proteins of Gαolf will likely not alter downstream signaling sufficiently. Activators of adenylate cyclase to boost cAMP levels itself may provide therapeutic benefit, but keeping the timing of cAMP production in sync with upstream neurotransmitter release will be challenging and may limit therapeutic efficacy [55]. Similarly, increasing cAMP through inhibiting its metabolism through phosphodiesterase may provide therapeutic efficacy, but may not allow for precise control of cAMP production in response to upstream neurotransmitter signaling [56]. Targeting of downstream effectors of cAMP produced by Gαolf such as downstream targets of protein kinase A and cyclic nucleotide gated channels also possible [57, 58], but further understanding of how these targets are altered in pre-clinical models of GNAL dystonia will be necessary.
Discussion
Mutations in GNAL are associated with both sporadic and familial dystonia [8]. These mutations lead to an adult onset dystonia that is usually not co-morbid with other disorders [1, 8]. This and other clinical characteristics of GNAL linked dystonia, suggest that this form of genetic dystonia may possibly have significant clinical and pathological overlap with idiopathic dystonia. This may represent a unique opportunity within dystonia research to utilize GNAL linked dystonia both in pre-clinical research and clinical research to determine, define and delineate the brain nuclei, cell types, and molecular events that are disturbed to cause disease. Translationally, this may represent unique opportunities for the design of rationale therapeutic strategies for a disease with few efficacious, well tolerated, or non-invasive therapies. Current animal models of GNAL linked dystonia have shown both striatal and cerebellar abnormalities, and a possible important role for striatal cholinergic signaling as a consequence of loss of Gαolf [45, 47, 59]. However, lack of overt dystonic symptoms in animal models of GNAL dystonia have prevented directly tying electrophysiological and biochemical endophenotypes with expression of dystonic motor phenotypes, and resulting in uncertainty over if these pathways are necessary or sufficient to drive dystonic motor phenotypes. Additionally, there are several open questions over the circuits, neuron types, and molecular signaling pathways that are altered to cause dystonia in these model systems. Generation of humanized mice that express the patient specific mutant forms of Gαolf or floxed mice that will allow for the conditional removal of Gαolf in specific cell types may allow for the development of symptomatic mice to directly address these questions. Further dissection of the mechanisms behind how mutations in GNAL lead to dystonia hold promise as a platform not only to mechanistically understand dystonia, but also as a platform to develop and test new anti-dystonic therapies.
Author contributions
MSM designed, conceptualized, and wrote the manuscript.
Funding
The author(s) declare(s) that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We would like to thank the National Institute of Health for funding this work through R00NS110878, and the Dystonia Medical Research Foundation for the original grant that started our GNAL linked dystonia research.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Fuchs, T, Saunders-Pullman, R, Masuho, I, Luciano, MS, Raymond, D, Factor, S, et al. Mutations in GNAL cause primary torsion dystonia. Nat Genet (2013) 45(1):88–92. doi:10.1038/ng.2496
2. Carecchio, M, Panteghini, C, Reale, C, Barzaghi, C, Monti, V, Romito, L, et al. Novel GNAL mutation with intra-familial clinical heterogeneity: expanding the phenotype. Parkinsonism Relat Disord (2016) 23:66–71. doi:10.1016/j.parkreldis.2015.12.012
3. Deutschländer, AB, and Wszolek, ZK. DYT-GNAL. In: MP Adam, GM Mirzaa, RA Pagon, SE Wallace, LJH Bean, and KW Gripp, editors. GeneReviews(®). University of Washington, Seattle: Seattle (1993).
4. Dos Santos, CO, Masuho, I, da Silva-Júnior, FP, Barbosa, ER, Silva, SM, Borges, V, et al. Screening of GNAL variants in Brazilian patients with isolated dystonia reveals a novel mutation with partial loss of function. J Neurol (2016) 263(4):665–8. doi:10.1007/s00415-016-8026-2
5. Ma, LY, Wang, L, Yang, YM, and Wan, XH. Mutations in GNAL gene in 214 cases with isolated dystonia. Parkinsonism Relat Disord (2015) 21(11):1367–8. doi:10.1016/j.parkreldis.2015.08.026
6. Masuho, I, Fang, M, Geng, C, Zhang, J, Jiang, H, Özgul, RK, et al. Homozygous GNAL mutation associated with familial childhood-onset generalized dystonia. Neurol Genet (2016) 2(3):e78. doi:10.1212/NXG.0000000000000078
7. Miao, J, Wan, XH, Sun, Y, Feng, JC, and Cheng, FB. Mutation screening of GNAL gene in patients with primary dystonia from Northeast China. Parkinsonism Relat Disord (2013) 19(10):910–2. doi:10.1016/j.parkreldis.2013.05.011
8. Vemula, SR, Puschmann, A, Xiao, J, Zhao, Y, Rudzińska, M, Frei, KP, et al. Role of Gα(olf) in familial and sporadic adult-onset primary dystonia. Hum Mol Genet (2013) 22(12):2510–9. doi:10.1093/hmg/ddt102
9. Dobričić, V, Kresojević, N, Westenberger, A, Svetel, M, Tomić, A, Ralić, V, et al. De novo mutation in the GNAL gene causing seemingly sporadic dystonia in a Serbian patient. Mov Disord : official J Mov Disord Soc (2014) 29(9):1190–3. doi:10.1002/mds.25876
10. Dufke, C, Sturm, M, Schroeder, C, Moll, S, Ott, T, Riess, O, et al. Screening of mutations in GNAL in sporadic dystonia patients. Mov Disord : official J Mov Disord Soc (2014) 29(9):1193–6. doi:10.1002/mds.25794
11. LeDoux, MS, Vemula, SR, Xiao, J, Thompson, MM, Perlmutter, JS, Wright, LJ, et al. Clinical and genetic features of cervical dystonia in a large multicenter cohort. Neurol Genet (2016) 2(3):e69. doi:10.1212/NXG.0000000000000069
12. Masuho, I, Chavali, S, Muntean, BS, Skamangas, NK, Simonyan, K, Patil, DN, et al. Molecular deconvolution platform to establish disease mechanisms by surveying GPCR signaling. Cell Rep (2018) 24(3):557–68. doi:10.1016/j.celrep.2018.06.080
13. Syrovatkina, V, Alegre, KO, Dey, R, and Huang, XY. Regulation, signaling, and physiological functions of G-proteins. J Mol Biol (2016) 428(19):3850–68. doi:10.1016/j.jmb.2016.08.002
14. Sassone-Corsi, P. The cyclic AMP pathway. Cold Spring Harbor Perspect Biol (2012) 4(12):a011148. doi:10.1101/cshperspect.a011148
15. Corvol, JC, Studler, JM, Schonn, JS, Girault, JA, and Hervé, D. Galpha(olf) is necessary for coupling D1 and A2a receptors to adenylyl cyclase in the striatum. J Neurochem (2001) 76(5):1585–8. doi:10.1046/j.1471-4159.2001.00201.x
16. Hervé, D, Le Moine, C, Corvol, JC, Belluscio, L, Ledent, C, Fienberg, AA, et al. Galpha(olf) levels are regulated by receptor usage and control dopamine and adenosine action in the striatum. J Neurosci (2001) 21(12):4390–9. doi:10.1523/JNEUROSCI.21-12-04390.2001
17. Zhuang, X, Belluscio, L, and Hen, R. G(olf)alpha mediates dopamine D1 receptor signaling. J Neurosci (2000) 20(16):Rc91. doi:10.1523/JNEUROSCI.20-16-j0001.2000
18. Drinnan, SL, Hope, BT, Snutch, TP, and Vincent, SR. G(olf) in the basal ganglia. Mol Cell neurosciences (1991) 2(1):66–70. doi:10.1016/1044-7431(91)90040-u
19. Belluscio, L, Gold, GH, Nemes, A, and Axel, R. Mice deficient in G(olf) are anosmic. Neuron (1998) 20(1):69–81. doi:10.1016/s0896-6273(00)80435-3
20. Khan, MM, Xiao, J, Hollingsworth, TJ, Patel, D, Selley, DE, Ring, TL, et al. Gnal haploinsufficiency causes genomic instability and increased sensitivity to haloperidol. Exp Neurol (2019) 318:61–70. doi:10.1016/j.expneurol.2019.04.014
21. Karlsson, M, Zhang, C, Méar, L, Zhong, W, Digre, A, Katona, B, et al. A single-cell type transcriptomics map of human tissues. Sci Adv (2021) 7(31):eabh2169. doi:10.1126/sciadv.abh2169
22. Sjöstedt, E, Zhong, W, Fagerberg, L, Karlsson, M, Mitsios, N, Adori, C, et al. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science (New York, NY) (2020) 367(6482):eaay5947. doi:10.1126/science.aay5947
23. Uhlén, M, Fagerberg, L, Hallström, BM, Lindskog, C, Oksvold, P, Mardinoglu, A, et al. Proteomics. Tissue-based map of the human proteome. Science (New York, NY) (2015) 347(6220):1260419. doi:10.1126/science.1260419
24. Krim, E, Aupy, J, Clot, F, Bonnan, M, Burbaud, P, and Guehl, D. Mutation in the GCH1 gene with dopa-responsive dystonia and phenotypic variability. Neurol Genet (2018) 4(2):e231. doi:10.1212/NXG.0000000000000231
25. Hoffmann, GF, Assmann, B, Bräutigam, C, Dionisi-Vici, C, Häussler, M, de Klerk, JB, et al. Tyrosine hydroxylase deficiency causes progressive encephalopathy and dopa-nonresponsive dystonia. Ann Neurol (2003) 54(Suppl. 6):S56–65. doi:10.1002/ana.10632
26. Hanihara, T, Inoue, K, Kawanishi, C, Sugiyama, N, Miyakawa, T, Onishi, H, et al. 6-Pyruvoyl-tetrahydropterin synthase deficiency with generalized dystonia and diurnal fluctuation of symptoms: a clinical and molecular study. Mov Disord : official J Mov Disord Soc (1997) 12(3):408–11. doi:10.1002/mds.870120321
27. Froukh, T. Genetic study in a family with dopa-responsive dystonia revealed a novel mutation in sepiapterin reductase gene. Pakistan J Med Sci (2019) 35(6):1736–9. doi:10.12669/pjms.35.6.1181
28. Weissbach, A, Pauly, MG, Herzog, R, Hahn, L, Halmans, S, Hamami, F, et al. Relationship of genotype, phenotype, and treatment in dopa-responsive dystonia: MDSGene review. Mov Disord : official J Mov Disord Soc (2022) 37(2):237–52. doi:10.1002/mds.28874
29. Reid, KM, Steel, D, Nair, S, Bhate, S, Biassoni, L, Sudhakar, S, et al. Loss-of-Function variants in DRD1 in infantile parkinsonism-dystonia. Cells (2023) 12(7):1046. doi:10.3390/cells12071046
30. Groen, JL, Ritz, K, Warner, TT, Baas, F, and Tijssen, MAJ. DRD1 rare variants associated with tardive-like dystonia: a pilot pathway sequencing study in dystonia. Parkinsonism Relat Disord (2014) 20(7):782–5. doi:10.1016/j.parkreldis.2014.04.002
31. Ferrini, A, Steel, D, Barwick, K, and Kurian, MA. An update on the phenotype, genotype and neurobiology of ADCY5-related disease. Genotype Neurobiol ADCY5-Related Dis (2021) 36(5):1104–14. doi:10.1002/mds.28495
32. Salpietro, V, Perez-Dueñas, B, Nakashima, K, San Antonio-Arce, V, Manole, A, Efthymiou, S, et al. A homozygous loss-of-function mutation in PDE2A associated to early-onset hereditary chorea. Mov Disord : official J Mov Disord Soc (2018) 33(3):482–8. doi:10.1002/mds.27286
33. Mencacci, NE, Kamsteeg, EJ, Nakashima, K, R'Bibo, L, Lynch, DS, Balint, B, et al. De novo mutations in PDE10A cause childhood-onset chorea with bilateral striatal lesions. Am J Hum Genet (2016) 98(4):763–71. doi:10.1016/j.ajhg.2016.02.015
34. Khan, A, Molitor, A, Mayeur, S, Zhang, G, Rinaldi, B, Lannes, B, et al. A homozygous missense variant in PPP1R1B/DARPP-32 is associated with generalized complex dystonia. Mov Disord : official J Mov Disord Soc (2022) 37(2):365–74. doi:10.1002/mds.28861
35. Corvol, JC, Muriel, MP, Valjent, E, Féger, J, Hanoun, N, Girault, JA, et al. Persistent increase in olfactory type G-protein alpha subunit levels may underlie D1 receptor functional hypersensitivity in Parkinson disease. J Neurosci (2004) 24(31):7007–14. doi:10.1523/JNEUROSCI.0676-04.2004
36. Goto, S. Striatal gαolf/cAMP signal-dependent mechanism to generate levodopa-induced dyskinesia in Parkinson’s disease. Front Cel Neurosci. (2017) 11:364. doi:10.3389/fncel.2017.00364
37. Morigaki, R, Okita, S, and Goto, S. Dopamine-induced changes in Gαolf protein levels in striatonigral and striatopallidal medium spiny neurons underlie the genesis of l-DOPA-induced dyskinesia in parkinsonian mice. Front Cell Neurosci (2017) 11:26. doi:10.3389/fncel.2017.00026
38. Alcacer, C, Santini, E, Valjent, E, Gaven, F, Girault, J-A, and Hervé, D. Gαolf mutation allows parsing the role of cAMP-dependent and extracellular signal-regulated kinase-dependent signaling in <span class="sc">l-</span>3. 4-Dihydroxyphenylalanine-Induced Dyskinesia (2012) 32(17):5900–10. doi:10.1523/JNEUROSCI.0837-12.2012
39. Schwindinger, WF, Betz, KS, Giger, KE, Sabol, A, Bronson, SK, and Robishaw, JD. Loss of G protein gamma 7 alters behavior and reduces striatal alpha(olf) level and cAMP production. J Biol Chem (2003) 278(8):6575–9. doi:10.1074/jbc.M211132200
40. Logothetis, DE, Kurachi, Y, Galper, J, Neer, EJ, and Clapham, DE. The beta gamma subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature (1987) 325(6102):321–6. doi:10.1038/325321a0
41. Ikeda, SR. Voltage-dependent modulation of N-type calcium channels by G-protein beta gamma subunits. Nature (1996) 380(6571):255–8. doi:10.1038/380255a0
42. Gulati, S, Jin, H, Masuho, I, Orban, T, Cai, Y, Pardon, E, et al. Targeting G protein-coupled receptor signaling at the G protein level with a selective nanobody inhibitor. Nat Commun (2018) 9(1):1996. doi:10.1038/s41467-018-04432-0
43. Clapham, DE, and Neer, EJ. G protein beta gamma subunits. Annu Rev Pharmacol Toxicol (1997) 37:167–203. doi:10.1146/annurev.pharmtox.37.1.167
44. Mamaligas, AA, and Ford, CP. Spontaneous synaptic activation of muscarinic receptors by striatal cholinergic neuron firing. Neuron (2016) 91(3):574–86. doi:10.1016/j.neuron.2016.06.021
45. Pelosi, A, Menardy, F, Popa, D, Girault, JA, and Hervé, D. Heterozygous gnal mice are a novel animal model with which to study dystonia pathophysiology. J Neurosci (2017) 37(26):6253–67. doi:10.1523/JNEUROSCI.1529-16.2017
46. Yu-Taeger, L, Ott, T, Bonsi, P, Tomczak, C, Wassouf, Z, Martella, G, et al. Impaired dopamine- and adenosine-mediated signaling and plasticity in a novel rodent model for DYT25 dystonia. Neurobiol Dis (2020) 134:104634. doi:10.1016/j.nbd.2019.104634
47. Aïssa, HB, Sala, RW, Georgescu Margarint, EL, Frontera, JL, Varani, AP, Menardy, F, et al. Functional abnormalities in the cerebello-thalamic pathways in a mouse model of DYT25 dystonia. eLife (2022) 11:e79135. doi:10.7554/eLife.79135
48. Moehle, MS, and Conn, PJ. Roles of the M4 acetylcholine receptor in the basal ganglia and the treatment of movement disorders. Mov Disord (2019) 34(8):1089–99. doi:10.1002/mds.27740
49. Chambers, NE, Millett, M, and Moehle, MS. The muscarinic M4 acetylcholine receptor exacerbates symptoms of movement disorders. Biochem Soc Trans (2023) 51(2):691–702. doi:10.1042/BST20220525
50. Moehle, MS, Pancani, T, Byun, N, Yohn, SE, Wilson, GH, Dickerson, JW, et al. Cholinergic projections to the substantia nigra pars reticulata inhibit dopamine modulation of basal ganglia through the M(4) muscarinic receptor. Neuron (2017) 96(6):1358–72. doi:10.1016/j.neuron.2017.12.008
51. Moehle, MS, Bender, AM, Dickerson, JW, Foster, DJ, Qi, A, Cho, HP, et al. Discovery of the first selective M4 muscarinic acetylcholine receptor antagonists with in vivo antiparkinsonian and antidystonic efficacy. ACS Pharmacol Translational Sci (2021) 4(4):1306–21. doi:10.1021/acsptsci.0c00162
52. Burke, RE, Fahn, S, and Marsden, CD. Torsion dystonia: a double-blind, prospective trial of high-dosage trihexyphenidyl. Neurology (1986) 36(2):160–4. doi:10.1212/wnl.36.2.160
53. Fahn, S, Burke, R, and Stern, Y. Antimuscarinic drugs in the treatment of movement disorders. Prog Brain Res (1990) 84:389–97. doi:10.1016/s0079-6123(08)60922-x
54. Moehle, MS, Bender, AM, Dickerson, JW, Foster, DJ, Qi, A, Cho, HP, et al. Discovery of the first selective M4 muscarinic acetylcholine receptor antagonists with in vivo antiparkinsonian and antidystonic efficacy. ACS Pharmacol Translational Sci (2021) 4:1306–21. doi:10.1021/acsptsci.0c00162
55. Doyle, TB, Hayes, MP, Chen, DH, Raskind, WH, and Watts, VJ. Functional characterization of AC5 gain-of-function variants: impact on the molecular basis of ADCY5-related dyskinesia. Biochem Pharmacol (2019) 163:169–77. doi:10.1016/j.bcp.2019.02.005
56. Erro, R, Mencacci, NE, and Bhatia, KP. The emerging role of phosphodiesterases in movement disorders. Mov Disord : official J Mov Disord Soc (2021) 36(10):2225–43. doi:10.1002/mds.28686
57. Slika, H, Mansour, H, Nasser, SA, Shaito, A, Kobeissy, F, Orekhov, AN, et al. Epac as a tractable therapeutic target. Eur J Pharmacol (2023) 945:175645. doi:10.1016/j.ejphar.2023.175645
58. Santoro, B, and Shah, MM. Hyperpolarization-activated cyclic nucleotide-gated channels as drug targets for neurological disorders. Annu Rev Pharmacol Toxicol (2020) 60:109–31. doi:10.1146/annurev-pharmtox-010919-023356
Keywords: GNAL, dystonia, GNAL dystonia, mutations in GNAL, models of dystonia
Citation: Moehle MS (2024) Mechanisms of GNAL linked dystonia. Dystonia 3:12079. doi: 10.3389/dyst.2024.12079
Received: 20 September 2023; Accepted: 22 April 2024;
Published: 09 May 2024.
Edited by:
G. W. Gant Luxton, University of California, Davis, United StatesCopyright © 2024 Moehle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark S. Moehle, mark.moehle@ufl.edu, moehlelab@gmail.com