 Sanjay Pandey
Sanjay Pandey Navneesh Yadav3
Navneesh Yadav3- 1Department of Neurology, Govind Ballabh Pant Postgraduate Institute of Medical Education and Research, New Delhi, India
- 2Department of Neurology and Stroke Medicine, Amrita Institute of Medical Sciences, Faridabad, Delhi National Capital Region, India
- 3Department of Genetics, University of Delhi South Campus, New Delhi, India
- 4Department of Microbiology and Bioinformatics, Aravind Medical Research Foundation, Madurai, Tamil Nadu, India
- 5Department of Neurology, All India Institute of Medical Sciences, Bhopal, India
Background: The clinical differentiation between essential tremor plus (ETP) and dystonic tremor (DT) is challenging. This study aimed at the genetic diagnosis of ETP and DT.
Methods: Whole exome sequencing was performed on 50 probands (ETP = 25; DT = 25) and analysed to identify variants in known genes linked with dystonia and essential tremor plus phenotypes.
Results: We identified pathogenic/likely pathogenic variants [THAP1 (n = 1) and ANO3 (n = 1)] in two patients with DT. In addition, one DT patient had a variant of uncertain significance in FUS and four patients had benign variants [CIZ1 (n = 1), COL6A3 (n = 1), GCH1 (n = 1), TENM4 (n = 1)]. One patient with ETP was detected to have a variant of uncertain significance in TENM4 and five patients with ETP had benign variants [COL6A3 (n = 2), VPS16 (n = 1), TAF1 (n = 1), KMT2B (n = 1)].
Conclusion: Genetic studies may be in an important biomarker in differentiating patients with ET plus from DT which is challenging in a clinical setting.
Introduction
Tremor is the most common movement disorder and is defined as an involuntary, rhythmic, oscillatory movement of a body part [1]. In the recent classification, tremor syndromes have been classified in two axes [1]. A new terminology essential tremor plus (ET plus) was added for patients with features of essential tremor (ET) and additional neurological signs of uncertain significance such as questionable dystonia, questionable ataxia, and mild cognitive impairment. Dystonic tremor syndromes are tremor syndromes combining tremor and dystonia as the leading neurological signs. Considering a lack of diagnostic markers regarding the questionable neurological signs, differentiation of ET plus and DT is challenging and adds to diagnostic confusion [2, 3]. There are many genes associated with the dystonic tremor phenotype and tremor may be their only motor manifestation at the onset [4]. Further, patients with ET plus may develop hard signs and may fall into the category of combined tremor syndrome like a dystonic tremor [5–7]. The search for causal genes for ET is still ongoing [8, 9]. Exome studies have reported the association of several genes [FUS, TENM4, HTRA2, SCN11A, NOTCH2NLC, and CACNA1] with ET [5, 8]. But they have been reported in single families only and not in other populations, suggesting that they may be private polymorphisms [5].
Given the potential for a high diagnostic yield from whole exome sequencing, we used this method to screen individuals with ET plus and DT to determine whether there is a genetic overlap in patients with ET plus and DT. To our knowledge, this is the first study to use whole exome sequencing to investigate genetic causes of ET plus.
Materials and methods
Recruitment of patient samples
Patients with ET plus and DT were recruited at GIPMER, New Delhi, (a tertiary care teaching institute) after obtaining approval from the institutional ethics committee (IEC). Written informed consent was obtained from all participating individuals as per the IEC guidelines. A total of 50 consecutive patients (ET plus: 25; DT: 25) were evaluated and their detailed history and clinical information were recorded with the help of a pre-designed form. All patients were examined by two neurologists (SP, CSR). Diagnosis of ET plus and DT was made based on the recent consensus classification. We enrolled patients with ETP having dystonia as a soft sign. A tremor in a body part affected by dystonia was labelled as dystonic tremor (DT). Dystonia was labelled as questionable if there was discordance between the two examiners (S.P., CSR) regarding its presence. If dystonia and tremor were found in different body parts, this was called tremor associated with dystonia (TAWD). For age at disease onset and disease duration, mean ± SD was calculated for each group (ET plus and DT). The severity of tremor was assessed using the Tremor Research Group Essential Tremor Rating Scale (TETRAS). About 5 mL of peripheral blood was collected in EDTA vacutainers from all the subjects recruited in the study.
Genetic analysis
Genomic DNA (gDNA) was isolated from the blood sample using the routine phenol-chloroform method at the Genetics lab (BKT) after obtaining IEC clearance and used for whole exome sequencing.
Whole exome sequencing
Exome library preparations of gDNA were made using SureSelect Human All Exon V5+UTR kit (Agilent Technologies, California, United States); and paired-end sequencing was performed on a NovaSeq 6000 at a commercial facility (MedGenome Labs, Bengaluru, India). Raw data with Phred quality score >Q30 were analysed using bioinformatic protocols previously described [10]. Using a combined variant calling file (VCF) generated for all the samples, both single nucleotide variations and insertions/deletions were called and annotated using KGGSeq [11].
Data analysis
The analysis focussed on previously reported dystonia (DYT) genes (n = 20) and hereditary essential tremor (ETM) genes (n = 3) based on their presence in the OMIM database (listed in Supplementary Table S1). The other ET genes were not included as either there was no variant with CADD score >20 identified in HS1BP3 gene (ETM2) or were only loci with no specific causal gene (ETM3) or no single nucleotide variants were reported in ETM6. For variant prioritization, only novel and rare variants with global minor allele frequency (MAF ≤ 0.01) present in public databases including 1000G, dbSNP v141, NHLBI GO ESP, ExAC, DiscovEHR, and gnomAD browser were retained. Furthermore, among them, only protein disturbing variants with CADD score>20 (denotes the top 1% most deleterious substitutions in the human genome) were taken forward.
Data validation
All the novel variants identified in the prioritized dataset were confirmed by PCR-Sanger sequencing. PCR was done using DreamTaq Polymerase (Thermo Fisher Scientific #EP0705) with primers as per the manufacturer’s protocol; and later sequencing of the PCR fragments was carried out at the Central Instrumentation Facility, UDSC.
Statistical analysis
Clinical data were analyzed using the “Statistical Package for the Social Sciences (SPSS)” PC-23 version and “Fisher’s exact test” was used to compare variables between ET plus and DT groups. For rare-variant burden test in known DYT and ETM genes between 50 cases (25 ET plus + 25 DT) and 100 ethnicity-matched controls with no history of dystonia/tremor, SKAT-O was performed in Efficient and Parallelizable Association Container Toolbox software as previously described [12].
Results
Demographic details
A total of 25 ET plus and 25 DT patients with 28 males and 22 females were recruited in the study. Patients with ET plus compared to those with DT had significantly more positive family history [84.0% (21/25) vs. 48% (12/25); p = 0.007], higher mean age [48.64 ± 15.22 vs. 38.28 ± 16.69; p = 0.026] and longer duration of disease [8.52 ± 5.94 vs. 5.08 ± 4.66; p = 0.027] (Table 1).
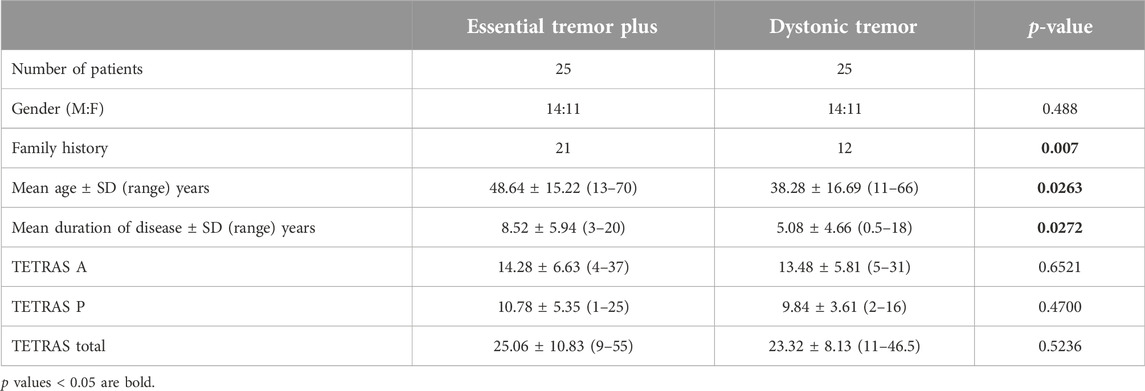
Table 1. Demographic and clinical characteristics of patients with essential tremor plus and dystonic tremor.
Clinical details
For 25 patients with ET plus, questionable dystonia was present in the neck (n = 14), or upper limb (n = 8). Laterocollis (n = 6), was the most common subtype of cervical dystonia present followed by torticpaut (n = 3), laterocaput (n = 2), retrocollis (n = 2), and anterocollis (n = 1). Three patients had questionable dystonia in the neck (retrocollis = 1; laterocollis = 1 and laterocollis + retocollis = 1) and upper limb.
Among 25 patients diagnosed with DT, the body distribution of dystonia was focal in 13, multifocal in 1, segmental in 8, and generalized in three patients. Focal dystonia was present in the upper limb (n = 9 including writer’s cramp in 7, eating dystonia in one and non-task specific dystonia in one patient), cranial (n = 2), cervical (n = 1), and trunk (n = 1). DT was present in 19 patients, TAD in five patients, and a combination of DT and TAD was present in only one patient. Detailed clinical information of all the study subjects has been provided in Supplementary Table S2.
Genetic analysis
Whole exome sequencing of 50 samples generated an average of ∼28 × 10 [6] reads per sample with an average %Q > 30 ∼95.05 (Supplementary Table S2) and ∼47.2 average mean coverage per sample. A total of 77,648 variants with MAF≤0.01 were called in the filtered dataset.
Novel and known variants with CADD >20 in DYT and ETM genes
We identified one heterozygous protein-coding rare variant each in two known autosomal dominant isolated dystonia genes, THAP1 (DYT-THAP1) and CIZ1 (DYT- CIZ1), and a novel variant c.979G>A in another autosomal dominant isolated dystonia gene, ANO3 (DYT- ANO3), which was confirmed by Sanger sequencing (Figure 1). All the identified variants had a high CADD score of >25 and among them, the c.416T>G variant in THAP1 and c.979G>A in ANO3 were predicted to be pathogenic by pathogenicity prediction tool, MetaRNN1 (Table 2). Of note, these two variants also had high conservation scores as evaluated by PhyloP100 which is based on multiple alignments of 99 vertebrate genome sequences to the human genome. On the other hand, the c.626G>A variant in CIZ1 was predicted to be benign with a low conservation score despite having a CADD score of 25.4 (Table 2).
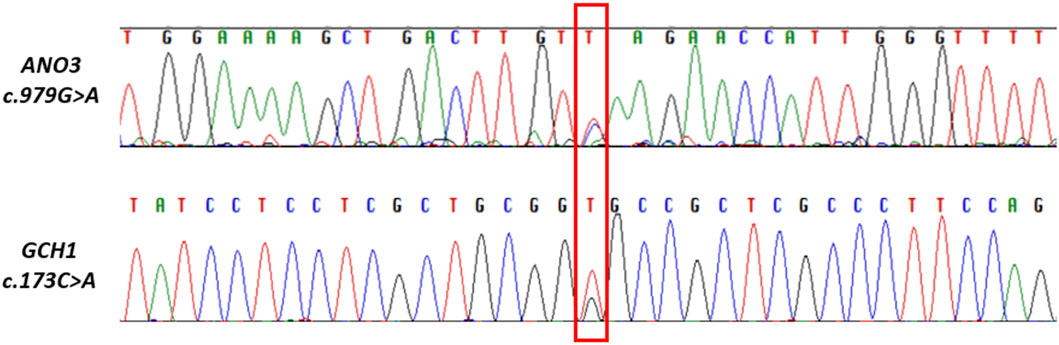
Figure 1. Electropherograms of Sanger sequencing showing part of ANO3 and GCH1gene sequences with novel variant (marked in red box), obtained using reverse primers.
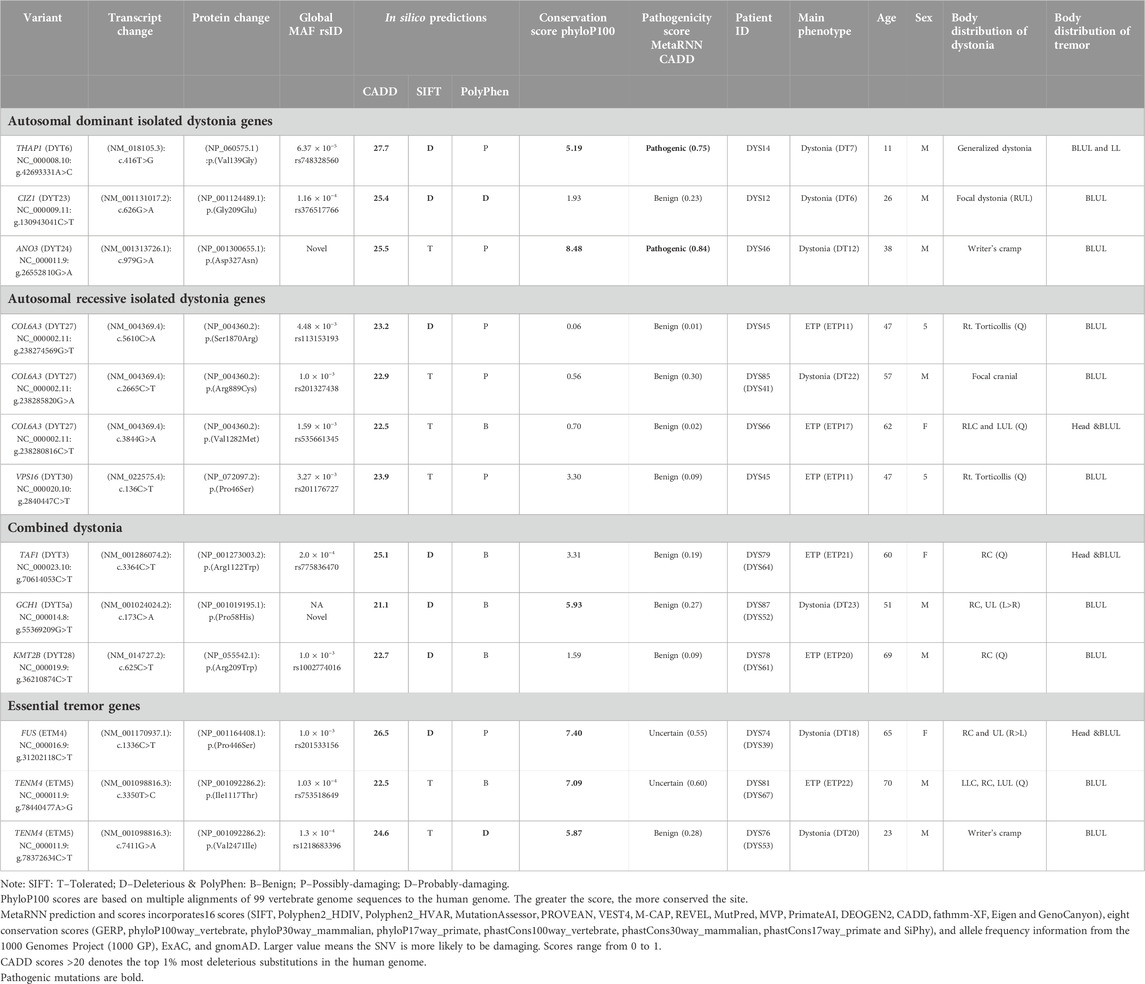
Table 2. In silico tools based pathogenicity prediction of the known or novel variants identified in known Dystonia and Essential Tremor genes in the study.
On screening for rare variants with CADD score>20 in known autosomal recessive (AR) isolated dystonia genes in the next step, we found three rare heterozygous variants in COL6A3 (DYT-COL6A3) and one rare heterozygous variant in VPS16 (DYT-VPS16), all predicted to be benign by MetaRNN (Table 2). Of note, all the variants identified in COL6A3 had a very low conservation score, while variant c.136C>T in VPS16 had a moderate score. Furthermore, we also identified one rare variant each in known combined dystonia genes, TAF1 (DYT/PARK-TAF1), GCH1 (DYT/PARK- GCH1); and KMT2B (DYT- KMT2B), all predicted to be benign by MetaRNN (Table 2). Of note, we identified a novel variant c.173C>A in GCH1, which was confirmed by Sanger sequencing (Figure 1) and which had a high conservation score as compared to the variants identified in TAF1 and KMT2B.
We tried levodopa in this patient and there was good improvement in hand dystonia. The neck dystonia required botulinum toxin injection.
For essential tremor genes, we identified one rare variant in FUS (ETM4) and two rare variants in TENM4 (ETM5) predicted to be variants of unknown significance and benign respectively by MetaRNN; but with a high conservation score (Table 2). Furthermore, SKAT-O analysis performed on rare variants in known DYT and ETM genes did not find any association (p-value≤0.05) between cases and controls possibly due to the limited sample size.
Genotype-phenotype correlations
Our study has provided some important observations.
Essential tremor plus
Of the six variants identified in this group, none were pathogenic. One patient had a variant of unknown significance in TENM4 and five had benign variants [COL6A3 (n = 2), VPS16 (n = 1), TAF1 (n = 1), KMT2B (n = 1)].
Dystonic tremor
Known or novel variants were observed in a total of seven DT patients but pathogenic/likely pathogenic known [THAP1 (n = 1) and novel ANO3 (n = 1)] variants were identified in only two patients.
THAP-1
A 24-year-old male (See Supplementary Video Case S1) presented with a 5-year history of abnormal movement of his body which started from his left hand. Gradually his movements were generalized and mainly involved the neck, upper limb, and trunk. On examination, he had generalized dystonia with more severe involvement of the neck, upper limb, and trunk (see Supplementary Video). He was treated with multiple sessions of injection botulinum toxin with a good response. His father had a similar history of generalized dystonia, but he did not consent to genetic testing.
ANO3
A 38-year-old male (See Supplementary Video Case S2) presented with 8-year history of task-specific focal hand dystonia in the form of writer’s cramp. He was working as a marketing executive where he had to write extensively to maintain the company records. His symptoms were insidious in onset and gradually progressive. On examination, he had primarily flexion type of writer’s cramp. He was injected with Ona botulinum toxin on multiple occasions with a good response to treatment. His father and uncle had a history of bilateral upper limbs postural tremor but they were not alive.
Discussion
The term ET-Plus was introduced in the last consensus classification [1]. It was defined as a tremor with the characteristics of essential tremor (ET) and additional neurological signs of uncertain significance such as questionable dystonic posturing. However, in the absence of a clear definition of questionable dystonia, there is a high rate of discordance among the experts regarding the diagnosis of ET plus and DT [2, 3]. We conducted this study to know the genetic profile of patients with ET plus and to establish overlap, if any, with DT patients. In this study, only two patients with DT had pathogenic mutations. One patient with ET plus and another with DT had a variant of unknown significance. As for ET genetics, our findings are consistent with the current understanding [5–9]. Although genetic component is likely to play an important role in the pathogenesis of ET with >50% of the affected individuals having a family history, very few disease-causing variants [DRD3, FUS, HTRA2, NOTCH2NLC, and TENM4] have been identified to date [8]. Of note, all the variants identified in DRD3 associated with ETM1 have been classified as variants of unknown significance suggesting the variants in this gene to be of minor significance. As for the FUS gene, the pathogenic variant is associated with both Amyotrophic lateral sclerosis type 6 and ETM4 suggesting the pleiotropic nature of this gene. Further, a pathogenic variant in the TENM4 gene is associated only with ETM5 (Supplementary Table S2). Besides their poor replicability in other studies, family members of patients with these variants have manifested phenotypes other than ET, like ataxia, parkinsonism, and autonomic dysfunction [5]. Genetic analyses of ET have been affected by different factors. In the absence of robust criteria many previous studies have included other tremor disorders misdiagnosed as ET, such as DT, spinocerebellar ataxias, and fragile X-associated tremor/ataxia syndrome (FXTAS). Furthermore, in DT, sometimes tremor can be the sole clinical manifestation and dystonia may appear later (e.g., DYT-ANO3) [4]. Genetic studies utilising sporadic or familial forms of ET and ET plus recruited following stringent diagnostic criteria are highly warranted to overcome these limitations.
There are some major limitations to this study including a small sample size. Also, many patients had a family history, they could not be tested due to the COVID pandemic at the time of patient recruitment. Further, WES is a powerful tool to identify all potential protein-coding genetic variants associated with a disease phenotype. However, it suffers from some limitations, such as a) low coverage efficiency: Some of the potential disease-causing variants may sometimes be missed owing to low coverage mostly due to poor DNA quality; library preparation, and/or some gene regions with repeat sequences. However, in our study, we obtained an average good quality data of ∼28 × 10 [6] reads per sample with an average %Q > 30 ∼95.05 (Supplementary Table S1) and ∼47.2 average mean coverage per sample; b) copy number variants: Detection of these structural variants in WES data has been challenging due to sophisticated bioinformatics tools which need to be used and sometimes the findings are not validated; and c) regulatory variants: Considering WES focuses on protein-coding variants, variants in non-coding regions of the gene which may have regulatory role on the gene expression are mostly not captured and likely missed in data interpretation. Despite these limitations, to the best of our knowledge, this is the first genetic study with a cohort of ET plus.
Conclusion
In our study, we identified pathogenic/likely pathogenic variants in two patients with DT, however, no pathogenic variants were identified in patients with ET plus. The findings of our study emphasize that genetic studies may be in an important biomarker in differentiating patients with ET plus from DT which may be challenging in a clinical setting.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving humans were approved by Institutional ethics committee, Maulana Azad Medical College, New Delhi. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
SP: Research project: Conception, organization, execution. Statistical analysis: Design, execution, review, and critique. Manuscript: Writing of the first draft, review, and critique. NY: Research project: Conception, Organization, Execution. Statistical analysis: Design, execution, review, and critique. Manuscript: Review, and critique. SD: Research project: Conception, organization, execution. Statistical analysis: Design, execution, review, and critique. Manuscript: Review, and critique. CR: Research project: Conception, organization, execution. Statistical analysis: Design, execution, review, and critique. Manuscript: Review, and critique. BT: Research project: Conception, organization, execution. Statistical analysis: Design, execution, review, and critique. Manuscript: Review, and critique.
Funding
The authors declare that financial support was received for the research, authorship, and/or publication of this article. This study was funded by grant # BT/PR26428/Med/12/783/2017 to SP and BT, from the Department of Biotechnology, New Delhi; junior research fellowship to SD, from project # BT/PR26428/Med/12/783/2017; and senior research fellowship to NY from #325541-2015, University Grants Commission, New Delhi.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontierspartnerships.org/articles/10.3389/dyst.2024.13181/full#supplementary-material.
SUPPLEMENTARY VIDEO CASE S1 | A 24-year-old male has generalized dystonia attributed to a mutation in THAP-1 gene. His father also has generalized dystonia.
SUPPLEMENTARY VIDEO CASE S2 | A 38-year-old male patient has right upper limb task-specific focal hand dystonia in the form of a writer's cramp attributed to a mutation in ANO3 gene.
Footnotes
1Available at https://varsome.com/
References
1. Bhatia, KP, Bain, P, Bajaj, N, Elble, RJ, Hallett, M, Louis, ED, et al. Consensus statement on the classification of tremors. From the task force on tremor of the international Parkinson and movement disorder society. Mov Disord (2018) 33(1):75–87. doi:10.1002/mds.27121
2. Pandey, S, and Bhattad, S. Questionable dystonia in essential tremor plus: a video-based assessment of 19 patients. Mov Disord Clin Pract (2019) 6:722–3. doi:10.1002/mdc3.12838
3. Pandey, S, Bhattad, S, and Hallett, M. The problem of questionable dystonia in the diagnosis of 'essential tremor-plus. Tremor Other Hyperkinet Mov (N Y) (2020) 10:27. PMID: 32864186. doi:10.5334/tohm.539
4. Pandey, S, Bhattad, S, and Dinesh, S. Tremor in primary monogenic dystonia. Curr Neurol Neurosci Rep (2021) 21(9):48. PMID: 34264428. doi:10.1007/s11910-021-01135-w
5. Pandey, S. Is essential tremor a family of diseases or a syndrome? A syndrome. Int Rev Neurobiol (2022) 163:31–59. Epub 2022 Mar 21. PMID: 35750367. doi:10.1016/bs.irn.2022.02.002
6. Lenka, A, and Pandey, S. Essential tremor: five new things. Neurol Clin Pract (2022) 12(2):183–6. PMID: 35747894; PMCID: PMC9208407. doi:10.1212/CPJ.0000000000001145
7. Lenka, A, and Pandey, S. Dystonia and tremor: do they have a shared biology? Int Rev Neurobiol (2023) 169:413–39. doi:10.1016/bs.irn.2023.04.002
8. Welton, T, Cardoso, F, Carr, JA, Chan, LL, Deuschl, G, Jankovic, J, et al. Essential tremor. Nat Rev Dis Primers (2021) 7(1):83. PMID: 34764294. doi:10.1038/s41572-021-00314-w
9. Clark, LN, Gao, Y, Wang, GT, Hernandez, N, Ashley-Koch, A, Jankovic, J, et al. Whole genome sequencing identifies candidate genes for familial essential tremor and reveals biological pathways implicated in essential tremor aetiology. EBioMedicine (2022) 85:104290. Epub 2022 Sep 29. PMID: 36183486; PMCID: PMC9525816. doi:10.1016/j.ebiom.2022.104290
10. John, J, Kukshal, P, Bhatia, T, Chowdari, KV, Nimgaonkar, VL, Deshpande, SN, et al. Possible role of rare variants in Trace amine associated receptor 1 in schizophrenia. Schizophr Res (2017) 189:190–5. doi:10.1016/j.schres.2017.02.020
11. Li, MX, Gui, HS, Kwan, JS, Bao, SY, and Sham, PC. A comprehensive framework for prioritizing variants in exome sequencing studies of Mendelian diseases. Nucleic Acids Res (2012) 40(7):e53. doi:10.1093/nar/gkr1257
Keywords: dystonic tremor, essential tremor plus, whole-exome sequencing, genotype, phenotype
Citation: Pandey S, Yadav N, Dinesh S, Rawat CS and Thelma BK (2024) Effort to differentiate essential tremor plus and dystonic tremor using whole exome sequencing: an exploratory study. Dystonia 3:13181. doi: 10.3389/dyst.2024.13181
Received: 25 April 2024; Accepted: 12 July 2024;
Published: 25 July 2024.
Edited by:
Aasef Shaikh, Case Western Reserve University, United StatesCopyright © 2024 Pandey, Yadav, Dinesh, Rawat and Thelma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sanjay Pandey, c2FuamF5c2dwZ2kyMDAyQHlhaG9vLmNvLmlu