 Azusa Ida
Azusa Ida Osamu Ishikawa1
Osamu Ishikawa1 Akihiko Uchiyama
Akihiko Uchiyama- 1Department of Dermatology, Ishii Hospital, Isesaki, Japan
- 2Department of Dermatology, Gunma University Graduate School of Medicine, Maebashi, Japan
- 3Department of Dermatology, Hirosaki University Graduate School of Medicine, Hirosaki, Japan
Dear Editors,
Dominant dystrophic epidermolysis bullosa pruriginosa (DDEB-Pr) is a rare subtype of dystrophic epidermolysis bullosa (DEB), characterized by severe itching and prurigo-like lesions [1]. Milia and toenail dystrophy are frequently observed as in other DEB subtypes. DDEB-Pr is caused by mutations in type VII collagen gene (COL7A1). We herein report a case of DDEB-Pr with a novel mutation in COL7A1.
A 28-year-old Japanese male was referred to our department with intractable prurigo-like lesions. He presented with numerous prurigo-like lesions on the entire lower legs, and some of them revealed erosions, milia and scar-like areas. Both toenails showed dystrophic change (Figures 1A, B). His symptoms developed around the age of 13, and have been refractory to topical steroid therapy. Medical interview disclosed that his father also had the same symptom. We performed a skin biopsy with a suspected diagnosis of DDEB-Pr. Histological examination revealed subepidermal cleft, dermal fibrosis and mild mononuclear cell infiltration in the upper to middle dermis (Figure 1C). Immunofluorescence staining with anti-type VII collagen antibody (LH7.2) showed decreased expression of type VII collagen in the lesional skin compared to that of normal control (Figure 1D).
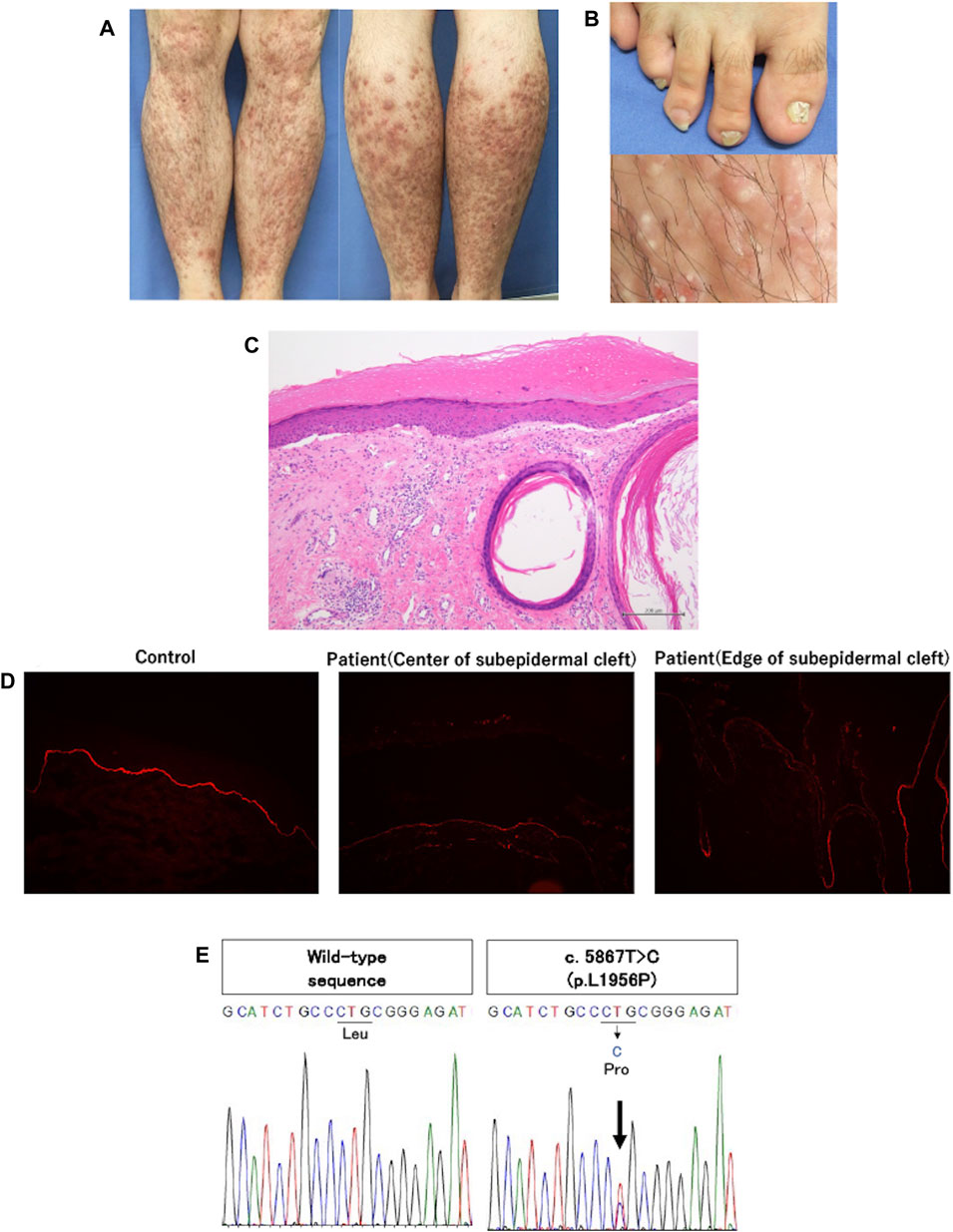
Figure 1. (A) Clinical features of the patient. Multiple brownish-reddish prurigo-like lesions were disseminated on both lower legs. Some of them were accompanied by erosions and hemorrhagic crusts, and scar-like lesion. (B) Clinical features of atrophic and deformed nails of bilateral big toes, and tiny white cysts, milia, in the prurigo-like lesions. (C) Histopathologic findings of prurigo-like lesion. Subepidermal cleft and milium were noted along with fibrosis and mild mononuclear cell infiltration in the upper to middle dermis. (D) Immunofluorescence study using anti-type VII collagen antibody (LH7.2). The expression of type VII collagen was slightly decreased at the edge of subepidermal cleft compared to healthy control, and was further decreased at the center of subepidermal cleft. (E) Direct sequencing of type VII collagen. c.5867T>C (p.L1956P) was detected in exon 73 of the patient (the right panel). The left panel shows wild-type sequences of healthy control.
After obtaining a written informed consent, genomic DNA extracted from his peripheral blood was subjected to direct sequencing of all exons and exon/intron junctions in COL7A1. His father did not receive the genetic analysis due to his personal reason. We identified a heterozygous mutation, c.5867T>C (p.L1956P) in exon 73 (NM_000094.4) (Figure 1E). In silico analyses, this mutation was “probably damaging” by Polyphen and “deleterious” by mutation tasting, and human genome variation database showed no same mutation in 1,208 healthy individuals. Based on the clinical and histological findings and laboratory results, we diagnosed him as DDEB-Pr.
There have been many reports on genetic mutations of DDEB-Pr. Glycine-substituted mutations are most common [2]. In addition, exon 87 skipping is thought to be associated with the phenotype of DDEB-Pr [3]. Although the mutation in exon 73 has not been reported so far, we assume that this mutation is pathogenic because it is located within 39 amino acids hinge region (p.1940-1978), which may serve as a crucial interaction site to other extracellular matrix [4].
The genotype-phenotype correlation has not been established in DDEB-Pr. There have been many reports of varying severity of clinical manifestations even among patients with the same mutation. We reported a novel mutation associated with the pruriginosa phenotype. We believe that this report may contribute to investigate the etiology of DDEB-Pr. It is assumed that severe itching and chronic skin damages by repeated scratching induce an inflammatory response and scar formation, which may result in the formation of prurigo-like lesions. Environmental, metabolic, immunological, hormonal and other factors have been postulated to be responsible for severe itching. In particular, the involvement of type 2 inflammation has been proposed in the pathogenesis of DDEB-Pr [5]. There have been several reports on the clinical efficacy of dupilumab for DDEB-Pr. Dupilumab blocks interleukin (IL)-4 and -13-mediated signaling pathway. IL-4 and -13 not only work as cytokine pruritogen but also could act as tissue remodeling and fibrogenic factors. In addition, IL-22 may be involved in the acanthosis. Thus, Th2 cell-mediated inflammation after blister formation could play certain roles in the formation of pruriginosa phenotype. However, the precise mechanism for clinical characteristics of DDEB-Pr remains to be elucidated.
This report adds new data to the database of genetic mutations. The genotype-phenotype correlation of DDEB-Pr is inconsistent, and further studies are warranted to understand the pathogenesis of DDEB-Pr.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ddbj.nig.ac.jp/, LC798144.
Ethics statement
Ethical approval was not required for the studies involving humans because this is a single case report. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from a by-product of routine care or industry. Written informed consent to participate in this study was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and the institutional requirements. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
OI contributed to the conception of the report, interpretation of the clinical laboratory findings, and drafting of the manuscript. AU and EA contributed to the acquisition and interpretation of the laboratory data. All authors contributed to the article and approved the submitted version.
Funding
The authors declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. McGrath, JA, Schofield, OM, and Eady, RA. Epidermolysis bullosa pruriginosa: dystrophic epidermolysis bullosa with distinctive clinicopathological features. Br J Dermatol (1994) 130:617–25. doi:10.1111/j.1365-2133.1994.tb13109.x
2. Kim, WB, Alavi, A, Walsh, S, Kim, S, and Pope, E. Epidermolysis bullosa pruriginosa: a systematic review exploring genotype-phenotype correlation. Am J Clin Dermatol (2015) 16:81–7. doi:10.1007/s40257-015-0119-7
3. Covaciu, C, Grosso, F, Pisaneschi, E, Zambruno, G, Gregersen, PA, Sommerlund, M, et al. A founder synonymous COL7A1 mutation in three Danish families with dominant dystrophic epidermolysis bullosa pruriginosa identifies exonic regulatory sequences required for exon 87 splicing. Br J Dermatol (2011) 165:678–82. doi:10.1111/j.1365-2133.2011.10414.x
4. Richer, BC, and Seeger, K. The hinge region of type VII collagen is intrinsically disordered. Matrix Biol (2014) 36:77–83. doi:10.1016/j.matbio.2014.04.006
Keywords: dystrophic epidermolysis bullosa pruriginosa, hinge region, novel mutation, COL7A1, genotype-phenotype correlation
Citation: Ida A, Ishikawa O, Uchiyama A and Akasaka E (2024) A case of dominant dystrophic epidermolysis bullosa pruriginosa with a novel mutation in the hinge region of COL7A1. J. Cutan. Immunol. Allergy 7:12844. doi: 10.3389/jcia.2024.12844
Received: 13 February 2024; Accepted: 19 April 2024;
Published: 01 May 2024.
Copyright © 2024 Ida, Ishikawa, Uchiyama and Akasaka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Azusa Ida, YXp1c2EuaWRhMjlAZ21haWwuY29t