 Abdualrahman Mohammed Abdualkader
Abdualrahman Mohammed Abdualkader Qutuba G. Karwi
Qutuba G. Karwi Gary D. Lopaschuk
Gary D. Lopaschuk Rami Al Batran
Rami Al Batran- 1Faculty of Pharmacy, Université de Montréal, Montréal, QC, Canada
- 2Montreal Diabetes Research Center, Montréal, QC, Canada
- 3Cardiometabolic Health, Diabetes and Obesity Research Network, Montréal, QC, Canada
- 4Division of BioMedical Sciences, Faculty of Medicine, Memorial University of Newfoundland, St. John’s, NL, Canada
- 5Mazankowski Alberta Heart Institute, University of Alberta, Edmonton, AB, Canada
Elevated levels of circulating branched-chain amino acids (BCAAs) and their associated metabolites have been strongly linked to insulin resistance and type 2 diabetes. Despite extensive research, the precise mechanisms linking increased BCAA levels with these conditions remain elusive. In this review, we highlight the key organs involved in maintaining BCAA homeostasis and discuss how obesity and insulin resistance disrupt the intricate interplay among these organs, thus affecting BCAA balance. Additionally, we outline recent research shedding light on the impact of tissue-specific or systemic modulation of BCAA metabolism on circulating BCAA levels, their metabolites, and insulin sensitivity, while also identifying specific knowledge gaps and areas requiring further investigation. Finally, we summarize the effects of BCAA supplementation or restriction on obesity and insulin sensitivity.
Introduction
Branched chain amino acids (BCAAs) are a group of three indispensable amino acids: leucine, isoleucine and valine. Together, they account for approximately 35% of the essential amino acids present in the human body. While the primary source of BCAAs is dietary intake [1], certain bacteria within the gut microbiome are capable of synthesizing them as well [2, 3]. However, the degree to which the gut microbiome produces BCAAs varies among individuals and is influenced by factors such as diet, gut microbiome composition, and overall health. Apart from serving as fundamental components in protein synthesis, BCAAs, especially leucine, play a critical role in stimulating protein synthesis through the activation of the mechanistic target of rapamycin (mTOR) signalling pathway [4]. Elevated plasma concentrations of BCAAs have been observed in both obese individuals and animal models of obesity [5–9]. Although plasma levels of other amino acid may also rise in obesity, the elevation in BCAAs are of particular interest because they appear to have unique effects in obesity-induced insulin resistance, and they are considered a major contributor to the pathology of type 2 diabetes (T2D) and coronary artery disease [10]. Numerous studies since the 1960s have consistently linked elevated plasma BCAAs with insulin resistance [7]. Furthermore, a landmark metabolomics profiling study even suggests that elevation in circulating BCAA levels can predict insulin resistance and T2D as much as 20 years prior to clinical presentation and a decade before any other known marker or test [11]. Interestingly, gastric bypass surgery in obese patients, which effectively lowers elevated BCAA levels, correlates with improved glucose homeostasis and enhanced insulin sensitivity [12]. Despite extensive research efforts, the underlying mechanisms by which elevated BCAA levels contribute to the development of insulin resistance and T2D remain unclear. In this Review, we highlight the major organs responsible for BCAA homeostasis. We then delve into how obesity and insulin resistance affect the communication between these organs, thereby influencing the maintenance of BCAA homeostasis. We also outline recent studies that sheds light on how modulating BCAA metabolism, either in a tissue-specific manner or at a whole-body level, impact circulating BCAA levels and their downstream metabolites, and the consequent effects on obesity and insulin resistance. We end by summarizing the effects of BCAA supplementation or restriction on obesity and insulin sensitivity.
Overview of BCAA catabolism
Plasma BCAA levels at the whole-body level are regulated by a delicate balance between input factors, such as dietary protein intake and proteolysis, and output factors, encompassing protein synthesis and oxidation. Insulin plays a pivotal role in maintaining this balance. Under normal and healthy conditions, insulin facilitates the cellular uptake of BCAAs while suppressing proteolysis, thus regulating plasma BCAA concentrations. However, in pathological states like insulin resistance, this regulatory mechanism may be disrupted. For instance, research on obese women has indicated that moderate obesity correlates with heightened proteolysis and impaired anti-proteolytic effects of insulin [13]. Another study suggested that the increased proteolysis observed in obesity and insulin resistance may be attributed to the compromised anti-proteolytic function of insulin [14]. The intricate regulatory relationship between insulin and BCAA metabolism has been extensively explored in previous literature reviews [15–17]. To facilitate a comprehensive understanding for the reader, we begin by providing essential information on BCAA catabolism and oxidation before delving into the role of BCAAs in mediating insulin resistance (Figure 1). The initial step in the BCAA catabolic pathway involves the reversible transamination of BCAAs catalyzed by branched-chain amino acid aminotransferase (BCAT). Notably, there exist two distinct isoforms of BCAT, namely, BCAT1 encoded by the cytosolic gene (Bcat1) and BCAT2 encoded by the mitochondrial gene (Bcat2). BCAT1 is the less common of the two isoforms and is primarily expressed in the cytoplasm, with a notable presence in the central and peripheral nervous systems [18, 19], while BCAT2 is the more ubiquitous isoform found in the mitochondria of most nonneuronal tissues, such as the heart, kidney, skeletal muscle and adipose tissue, excluding the liver [20, 21]. BCAT transfers the amino group from BCAAs to α-ketoglutarate, producing glutamate and the corresponding branched-chain α-keto acids (BCKAs): α-ketoisocaproate (KIC) from leucine, α-keto-β-methylvalerate (KMV) from isoleucine, and α-ketoisovalerate (KIV) from valine. This transamination reaction generates ammonia as a byproduct, particularly in the muscles. To remove excess ammonia, the muscle activates the alanine cycle (also known as the Cahill cycle), converting pyruvate to alanine by attaching the amino group from glutamate to pyruvate. Additionally, muscles convert glutamate and ammonia to glutamine as another means of ammonia detoxification. Both alanine and glutamine, as non-toxic carriers of ammonia, are transported to the liver, where the ammonia can be further processed and excreted [22, 23].
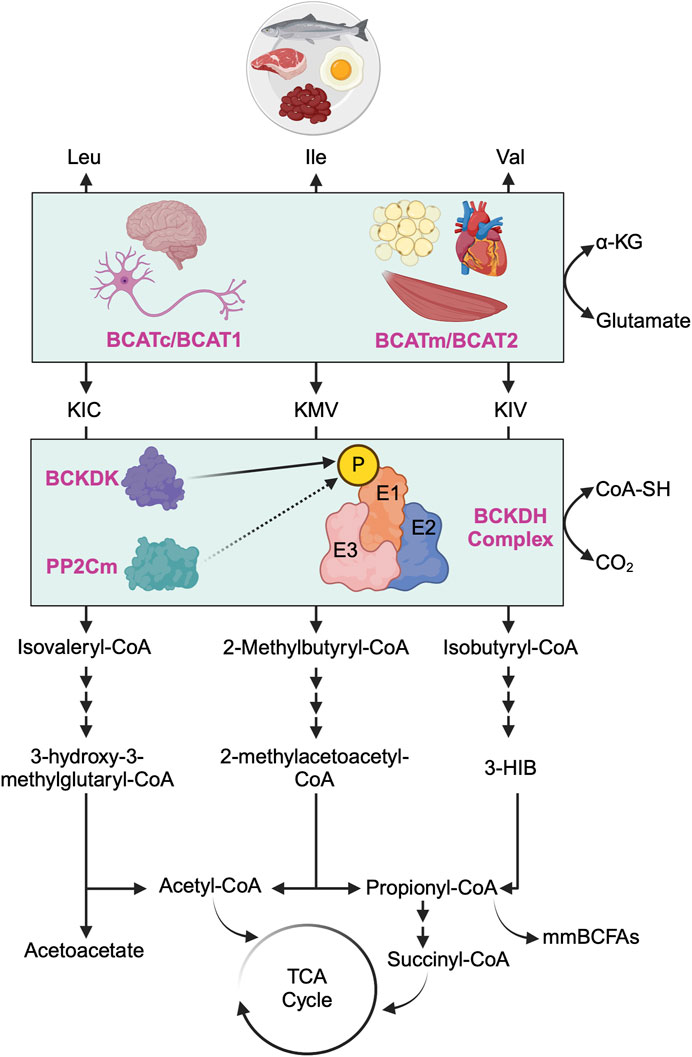
Figure 1. Overview of branched-chain amino acid catabolism pathway. The initial and shared step in the catabolism of all three branched-chain amino acids (BCAAs) - leucine (Leu), isoleucine (Ile), and valine (Val) - involves the reversible transamination of BCAAs to produce branched-chain alpha-ketoacids (BCKAs). Specifically, Leu yields α-ketoisocaproate (KIC), Ile yields α-keto-ß-methylvalerate (KMV), and Val yields α-ketovalerate (KIV). This transamination process is catalyzed by two distinct isoforms of branched-chain amino acid aminotransferase (BCAT): the cytosolic isoform (BCATc/BCAT1, encoded by the Bcat1 gene), predominantly found in the central nervous system and peripheral nerves, and the mitochondrial isoform (BCATm/BCAT2, encoded by the Bcat2 gene), primarily located in the mitochondria of most nonneuronal tissues. Subsequently, all three BCKAs (KIC, KMV, and KIV) undergo irreversible oxidative decarboxylation, facilitated by the branched-chain alpha-ketoacid dehydrogenase (BCKDH) complex, which serves as the rate-limiting enzyme in BCAA oxidation. The BCKDH complex comprises of three components: E1 (encoded by Bckdha and Bckdhb genes), E2 (encoded by Dbt gene), and E3 (encoded by Dld gene). The activity of the BCKDH complex is tightly regulated by BCKDH kinase (BCKDK), which phosphorylates E1 of the BCKDH complex and inhibits its activity (i.e., inhibiting BCAA oxidation), whereas protein phosphatase 2Cm (PP2Cm) dephosphorylates E1 of the BCKDH complex and activates its activity (i.e., activating BCAA oxidation). Post-decarboxylation, each BCKA follows a distinct metabolic pathway, generating acyl-CoA derivatives (isovaleryl-CoA from KIC, 2-methylbutyryl-CoA from KMV, and isobutyryl-CoA from KIV) and various downstream metabolites. These metabolites include critical metabolic intermediates for the TCA cycle, such as acetyl-CoA or succinyl-CoA, as well as acetoacetate, metabolic end products of Leu catabolism, 3-hydroxyisobutyrate (3-HIB), a downstream metabolite of Val that stimulates fatty acid uptake, and monomethyl branched-chain fatty acids (mmBCFAs), adipocyte-specific metabolites derived from mitochondrial BCAA catabolism, namely, propionyl-CoA.
Following BCAAs transamination, the irreversible oxidative decarboxylation of BCKAs is catalyzed by the branched-chain α-ketoacid dehydrogenase (BCKDH) complex, serving as the rate-limiting step in BCAA oxidation. The BCKDH complex comprises three components: E1 (encoded by Bckdha and Bckdhb genes, functioning as a thiamine-dependent decarboxylase), E2 (encoded by Dbt gene, functioning as dihydrolipoyl transacylase), and E3 (encoded by Dld gene, functioning as dihydrolipoamide dehydrogenase) [17]. The activity of the BCKDH complex is tightly regulated by BCKDH kinase (BCKDK), which phosphorylates and inhibits the BCKDH complex, and protein phosphatase 2Cm (PP2Cm), responsible for dephosphorylating and activating the BCKDH complex [24]. After decarboxylation, each BCKA follows a distinct metabolic route, ultimately leading to the formation of either acetyl-CoA or succinyl-CoA for energy production in the tricarboxylic acid (TCA) cycle or other metabolic intermediates such as acetoacetate, 3-hydroxyisobutyrate (3-HIB) or monomethyl branched-chain fatty acids (mmBCFAs).
Major organs responsible for BCAA homeostasis
BCAA metabolism is an intricate process that relies on inter-organ communication to maintain BCAA homeostasis. Among the key contributors to the circulating pool of BCAAs, skeletal muscle emerges as a predominant site. Skeletal muscle plays a pivotal role in BCAA transamination, primarily owing to the substantial abundance of BCAT2 within the muscle and its considerable muscle mass [19]. Importantly, skeletal muscle not only serves as a hub for BCAA transamination but also stands out as a major site for BCAA oxidation (accounting for 59% of whole-body BCAA oxidation) and protein synthesis (contributing to 24% of the total protein synthesis from BCAAs) [16]. In contrast, the liver does not engage in BCAA transamination or BCKA re-amination due to the lack of BCAT2 in hepatocytes [15]. Instead, owing to the liver’s high BCKDH activity, BCKAs derived from BCAA transamination in extrahepatic tissues are transported to the liver, where they can serve as substrates for BCAA oxidation [15, 25]. While BCKDH complex activity is notably high in the liver and comparatively low in adipose tissue [19, 26], recent tracing studies in mice have uncovered brown adipose tissue as an additional significant site for BCAA oxidation, constituting 19% of whole-body BCAA oxidation, followed by the liver at 8% [27]. This observation has been further supported by another study that used positron emission tomography-computed tomography scans with a leucine-analogue tracer in mice and humans. The study concluded that, upon cold exposure, brown adipose tissue, but not white adipose tissue, significantly contributes to systemic BCAA clearance by enhancing BCAA uptake in this tissue compartment to generate heat through thermogenesis [28].
Numerous studies have demonstrated that inter-organ communication essential for maintaining BCAA homeostasis is disturbed in obesity and insulin resistance. For example, studies showed that in two different rodent models of obesity and insulin resistance (ob/ob mice and Zucker rats), the BCKDH activity is decreased in the liver [29–31]. Additionally, other studies have consistently revealed reductions in BCKDH complex expression or activity in white adipose tissue across various models of obesity and insulin resistance [32, 33]. Remarkably, transplanting white adipose tissue from wild-type mice into BCAT2 or PP2CM deficient mice has been found to lower circulating BCAA levels [32, 34], highlighting the pivotal role of adipose tissue in regulating BCAA levels systemically. In line with these findings, Neinast and colleagues uncovered that in db/db mice, a model of severe insulin resistance, BCAA oxidation is impaired in adipose tissues and liver and redirected towards skeletal muscle [27]. The same group also demonstrated that excess BCAA oxidation in skeletal muscle leads to the secretion of 3-HIB, a downstream metabolite of valine, which, in turn, stimulates muscle fatty acid uptake and lipid accumulation, thereby exacerbating insulin resistance [35]. Another group hypothesized that in obesity and insulin resistance, the accumulation of C3 and C5 acylcarnitines in muscle, which are by-products of BCAA catabolism and markers of incomplete fat oxidation, may contribute to insulin resistance [36, 37]. A recent hypothesis posits a direct association between BCKAs and insulin resistance, where exposure of muscle cells to high concentrations of BCKAs results in the inhibition of insulin-induced AKT phosphorylation (also known as protein kinase B) and glucose uptake [38], indicating a direct role of BCKAs in impairing insulin signalling. Finally, the classical mechanism linking elevated BCAA levels with insulin resistance involves chronic hyperactivation of mTORC1 and its downstream effector, ribosomal protein S6 kinase 1 (S6K1), also known as p70-S6K. This hyperactivation phosphorylates and inhibits insulin receptor substrate 1 (IRS-1), thus blunting insulin signalling and contributing to insulin resistance [39–41].
Indeed, the role of the gut microbiome in maintaining BCAA homeostasis was historically overlooked due to the complexity of the microbiome, technological limitations, and a traditional focus on host genetics and diet. However, recent advances in this domain have highlighted the microbiome’s critical role in BCAA synthesis, regulation, and interaction with host metabolism. It is now evident that the gut microbiome contributes to the overall pool of BCAAs, potentially influencing the development of insulin resistance [42]. For example, a landmark study identified Bacteroides vulgatus and Prevotella copri as two key species of gut microbiome bacteria responsible for elevated BCAA biosynthesis and associated with insulin resistance in humans [3]. This study also demonstrated that Prevotella copri can induce insulin resistance, exacerbate glucose intolerance, and increase circulating levels of BCAAs in mice. Furthermore, a recent study demonstrated that feeding mice a variety of protein sources mirroring the composition of the Western diet exacerbates insulin resistance. This effect is attributed to an increase in gut microbial branched-chain fatty acids (BCFA) [43], a class of short-chain fatty acids produced in the gut through the proteolytic fermentation of BCAAs.
Nevertheless, in the subsequent sections, we will discuss and summarize the effects of modifying BCAA catabolism, either selectively in a tissue-specific manner (muscles, liver, adipose tissue, and heart) or systemically, on circulating BCAA levels and insulin sensitivity (Table 1)
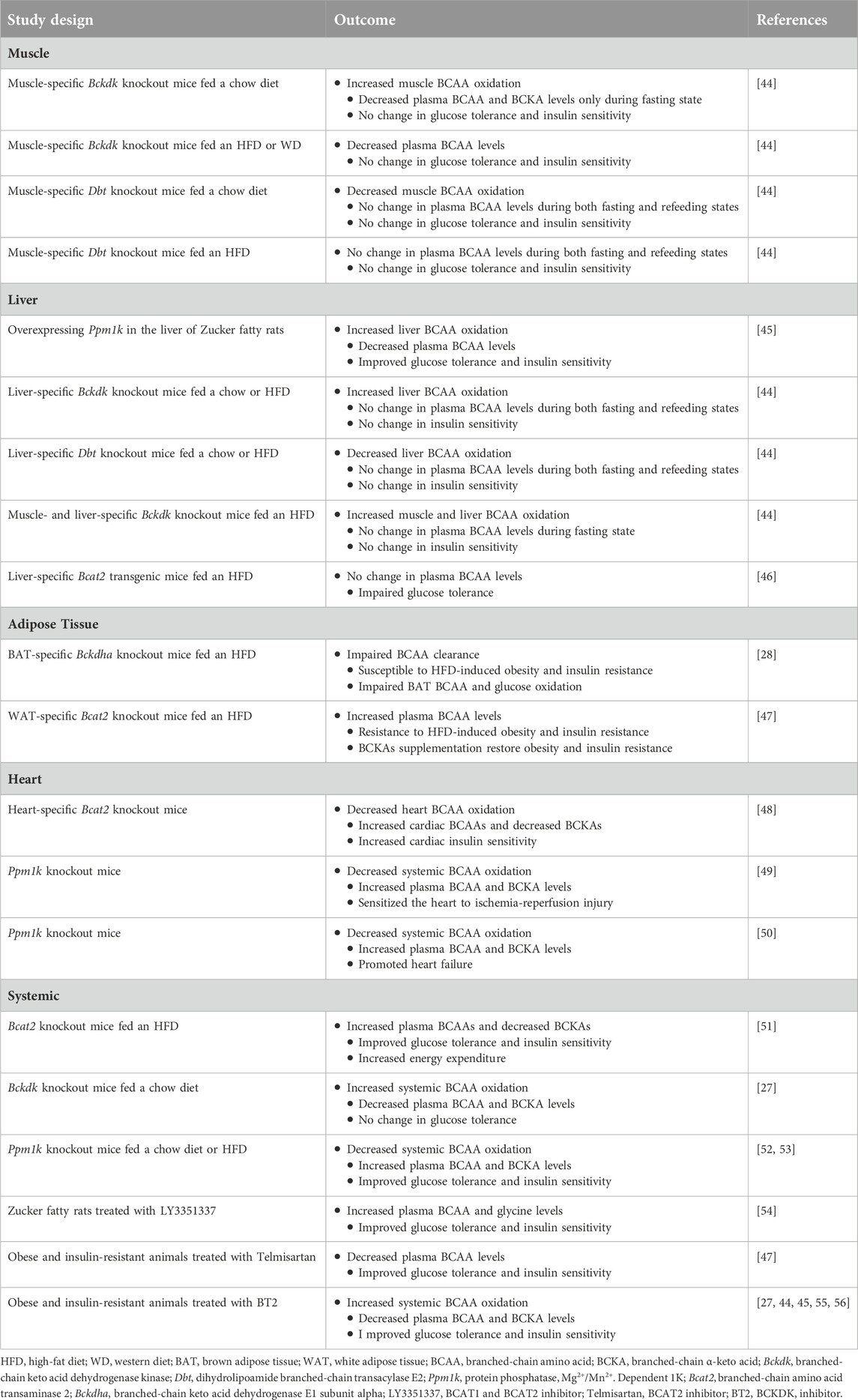
Table 1. The effects of modulating BCAA catabolism in various tissue compartments or systemically on insulin sensitivity in lean and obese animals.
Modulating BCAA catabolism to treat insulin resistance
Muscle: Skeletal muscle plays a crucial role in maintaining BCAA homeostasis, serving as the primary site for whole-body BCAA oxidation. In Zucker-fatty rats, BCKDH activity is elevated in skeletal muscle but reduced in the liver compared to Zucker-lean rats [29]. Similarly, Neinast and colleagues demonstrated that in db/db mice, but not in mice fed a high-fat diet for 14 weeks, BCAA oxidation is increased in skeletal muscle and decreased in the liver and adipose tissue [16, 27]. Furthermore, several studies have noted diminished BCAA oxidation in adipose tissues during obesity and insulin resistance [30, 32]. These collective observations from multiple research groups have led to the hypothesis that excess BCAA oxidation in skeletal muscle may contribute to insulin resistance. This may occur via two potential mechanisms: 1) through the overproduction of 3-HIB in muscle or 2) via the accumulation of acylcarnitines derived from muscle BCAA breakdown. In both scenarios, this would impair fatty acid oxidation and promote lipotoxicity [29, 37, 57]. To test this hypothesis, Blair et al [44] generated muscle-specific knockout mice lacking either the Bckdk gene, responsible for phosphorylating E1 of the BCKDH complex and inhibiting its activity, or the Dbt gene, crucial for BCAA oxidation as it encodes the E2 component of the BCKDH complex. Interestingly, their investigation revealed that augmenting muscle BCAA oxidation lowered plasma BCAA and BCKA levels only during the fasting state in muscle-specific Bckdk knockout mice fed a chow diet compared to their control littermates, human α-skeletal actin (HSA)-Cre mice. Conversely, diminishing muscle BCAA oxidation did not significantly alter plasma BCAA levels during both fasting and refeeding states in muscle-specific Dbt knockout mice fed a chow diet compared to HSA-Cre mice. However, the administration of a single bolus of BCAA resulted in impaired BCAA and BCKA clearance in muscle-specific Dbt knockout mice when compared to their controls. These findings indicate that manipulating muscle BCAA oxidation under healthy conditions impacts circulating BCAA levels predominantly during fasting.
To investigate whether modulating muscle BCAA oxidation impacts glucose homeostasis, muscle-specific Bckdk knockout mice were subjected to chronic feeding regimens of either a Western diet or a high-fat diet spanning from 4 up to 12 weeks, followed by assessment of insulin sensitivity and glucose handling using hyperinsulinemic-euglycemic clamp and glucose tolerance tests (GTT), respectively. Surprisingly, despite observing a reduction in plasma BCAAs and an increase in the 3-HIB/valine ratio during the fasted state in muscle-specific Bckdk knockout mice fed the obesogenic diet (Western diet or high-fat diet), this alteration did not manifest in changes in insulin sensitivity or glucose handling. Specifically, there were no discernible differences in euglycemic clamp and GTT outcomes between muscle-specific Bckdk knockout mice and their controls. Similarly, inhibiting muscle BCAA oxidation in muscle-specific Dbt knockout mice fed a high-fat diet did not affect insulin sensitivity during a euglycemic clamp or alter glucose handling during a GTT. Additionally, there were no significant changes observed in plasma BCAA levels or the 3-HIB/valine ratio in both fasted and refed states. These collective findings suggest that augmenting or diminishing muscle BCAA oxidation has no impact on whole-body insulin sensitivity in mice subjected to various obesogenic diets. While this study did not directly measure it, further exploration into the effects of modulating muscle BCAA oxidation on muscle insulin sensitivity itself would be intriguing. Moreover, investigating whether muscle acylcarnitine species, particularly C3 and C5, as well as BCKA levels play a role in improving or exacerbating muscle insulin sensitivity could provide valuable insights.
Liver: While the liver lacks the BCAT enzyme necessary for the conversion of BCAAs into BCKAs and vice versa, it remains a pivotal site for BCAA oxidation and protein synthesis, contributing up to 27% of whole-body BCAA incorporation into proteins [27]. As previously noted, multiple studies have demonstrated markedly elevated expression of liver BCKDK, which phosphorylates and inhibits BCKDH complex activity, in various models of obese and insulin-resistant rodents [29–31]. In an effort to understand whether the reduction in liver BCAA oxidation contributes to the development of insulin resistance, White and colleagues utilized adenovirus-mediated delivery of Ppm1k, the gene encoding PP2Cm (which dephosphorylates and activates the BCKDH complex), to specifically overexpress PP2Cm in the liver of Zucker fatty rats [45]. Their findings demonstrated that liver PP2Cm overexpression enhanced liver BCKDH activity, reduced circulating BCAAs, alleviated hepatic steatosis, and improved glucose tolerance and insulin sensitivity. Interestingly, hepatic overexpression of PP2Cm increased the phosphorylation of ATP-citrate lyase (ACLY), a critical enzyme involved in lipid synthesis. This activation of ACLY subsequently stimulated de novo lipogenesis, thereby integrating BCAA metabolism with lipid metabolism.
On the contrary, manipulating liver BCAA oxidation levels through targeting either the Bckdk or Dbt gene in the liver, using an adeno-associated viral (AAV) vector carrying the Cre recombinase gene under the control of the thyroxine-binding globulin (TBG) promoter (AAV8-TBG-Cre) in Bckdk or Dbt floxed mice, did not influence circulating BCAA levels in either fasted or refed states, regardless of whether the mice were subjected to a chow or high-fat diet for 4–5 weeks [44]. Furthermore, neither enhancing nor suppressing liver BCAA oxidation affected whole-body insulin sensitivity in mice fed a normal chow or high-fat diet. Of note, augmenting both muscle and liver BCAA oxidation in mice, achieved by treating muscle-specific Bckdk knockout mice with AAV8-TBG-Cre to generate double knockout mice, also failed to impact whole-body insulin sensitivity in mice subjected to a high-fat diet for 6 weeks. This was observed despite a notable reduction in fasting plasma BCAAs and an increase in the 3-HIB/valine ratio in BCKDK double knockout mice compared to their control counterparts. Although the reasons for the discrepancy between the results regarding the manipulation of liver BCAA oxidation and insulin sensitivity in the mouse and rat studies remain unclear, it has been suggested that species differences may account for the contrasting outcomes [57]. Of note, a recent study revealed that mice lacking PP2Cm globally are protected against high-fat-diet-induced insulin resistance. Interestingly, this investigation also demonstrated that BCKAs selectively inhibits the mitochondrial pyruvate carrier (MPC) in hepatocytes, thus suppressing gluconeogenesis from pyruvate [52]. Nevertheless, further research is warranted to delineate the role of liver BCAA catabolism in insulin resistance.
Adipose tissue: Adipose tissue, traditionally viewed as a passive site for energy storage, is now recognized as a dynamic regulator impacting various aspects of whole-body metabolism, including BCAA catabolism. In conditions like obesity and insulin resistance, there is a notable suppression in the expression of nearly all enzymes responsible for BCAA catabolism, particularly within white adipose tissue [30, 32, 55, 58]. This decrease in BCAA catabolism is considered a significant contributor to the systemic elevation of BCAA levels during obesity and insulin resistance [33, 59]. Cross-tissue flux studies comparing lean and healthy individuals to insulin-sensitive or insulin-resistant obese subjects revealed negligible uptake of BCAAs from human abdominal subcutaneous white adipose tissue [33]. However, BCAA catabolic enzyme levels were markedly reduced in omental fat, a specific type of visceral fat, but not in subcutaneous white adipose tissue of obese individuals with metabolic syndrome compared to weight-matched healthy obese subjects. This finding suggests that alterations in BCAA catabolism in visceral white adipose tissue significantly contribute to the BCAA metabolic phenotype in individuals with insulin resistance. Furthermore, adipose tissue not only utilizes BCAAs to support adipocyte differentiation and lipogenesis [60], but it also has the capacity to release adipocyte-specific metabolites stemming from mitochondrial BCAA catabolism, such as mmBCFAs. These metabolites play a role in fueling de novo lipogenesis, with their levels being notably decreased in obese animals and increased during prolonged fasting [61]. Consequently, it is tempting to speculate that the reduced levels of plasma mmBCFAs observed in obesity may be attributed to decreased BCAA catabolism within this specific tissue compartment.
Adipose tissue, particularly brown adipose tissue, plays a significant role in utilizing BCAAs for thermogenesis during cold exposure in both mice and humans. This process contributes to systemic BCAA clearance by enhancing BCAA uptake via SLC25A44, a mitochondrial BCAA transporter [28]. Notably, BCAA clearance following oral administration of BCAAs is compromised in mice with targeted deletion of Bckdha in brown adipose tissue, the gene responsible for encoding the E1 component of the BCKDH complex and critical for BCAA oxidation. Moreover, brown adipose tissue-specific Bckdha knockout mice showed increased susceptibility to high-fat diet-induced obesity and insulin resistance, coupled with impaired glucose oxidation within brown adipose tissue. These findings underscore the critical role of intact BCAA oxidation in brown adipose tissue for systemic BCAA clearance and the amelioration of obesity and insulin resistance. Conversely, a recent study indicates that white adipose tissue-specific Bcat2 knockout mice display resistance to high-fat diet-induced obesity and insulin resistance, attributed to enhanced browning and thermogenesis in white adipose tissue [47]. Intriguingly, the study also revealed that BCKAs inhibits white adipose tissue browning through the acetylation of the PR domain-containing protein 16 (PRDM16). Furthermore, supplementation of BCKAs in white adipose tissue-specific Bcat2 knockout mice reverses these favorable effects, leading to the reinstatement of obesity and insulin resistance. These findings suggest that mitigating BCAA transamination into BCKAs in white adipose tissue, consequently affecting BCAA oxidation in this tissue compartment, is beneficial in attenuating obesity and insulin resistance.
One pivotal question emerges from the findings of these two studies: Why is the suppression of BCAA oxidation in brown adipose tissue detrimental rather than protective against insulin resistance, whereas its suppression in white adipose tissue appears to confer a protective effect? One potential explanation for this phenomenon is that inhibiting BCAA oxidation in brown adipose tissue not only raises BCAA levels but also elevates the level of BCKAs, thereby triggering insulin resistance. Conversely, targeting BCAT2 in white adipose tissue leads to increased BCAA levels while concurrently reducing BCKAs, thus mitigating insulin resistance. In support of this hypothesis, mice with whole-body Bcat2 deletion exhibit elevated plasma BCAAs and decreased BCKAs, yet remained protected from high-fat diet-induced obesity and insulin resistance [51]. Remarkably, this protection persists even in the presence of mTORC1 hyperactivation, as evidenced by the phosphorylation of mTOR downstream targets such as eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1) and S6K1 in the gastrocnemius of Bcat2 knockout fasted mice. Collectively, these findings suggest that it may not be the mere elevation of circulating BCAAs per se that drives insulin resistance, but rather the accumulation of BCKAs that plays a crucial role in mediating insulin resistance. Given that skeletal muscle predominantly facilitates the conversion of BCAAs into BCKAs, it would be interesting to explore whether decreasing BCKAs, particularly in muscle tissue, by targeting BCAT2 yields outcomes akin to reducing BCKAs in white adipose tissue. Future studies are imperative to answer this question.
Heart: Decreased cardiac BCAA oxidation has been linked to the development of cardiac insulin resistance and impaired cardiac insulin signalling pathways [62]. Direct measurement of cardiac BCAA oxidation rates in isolated working mouse hearts demonstrated that these rates are decreased in a mouse model of high-fat diet-induced obesity [63]. This is associated with a decreased activity of Akt and glycogen synthase kinase-3β (GSK-3β) and cardiac insulin-stimulated glucose oxidation rates in obese mice [63]. Similarly, cardiac BCAA oxidation rates are also decreased in the failing heart, which is associated with impaired insulin signalling and insulin-stimulated glucose oxidation rates [64]. A whole-body PP2Cm deletion, a maneuver which decreases the activity of BCKDH complex and BCAA oxidation, is associated with decreased glucose oxidation by inhibiting pyruvate dehydrogenase (PDH) activity and increased vulnerability to myocardial ischemia/reperfusion injury [49].
Since impaired BCAA oxidation leads to the accumulation of BCAAs and BCKAs, it is difficult to ascertain whether BCAAs or BCKAs contribute to cardiac insulin resistance. Selective increasing cardiac BCKA levels abrogates insulin-stimulated cardiac glucose oxidation rates via inhibiting insulin signalling pathway ex vivo [48]. While BCKAs could be re-aminated to their correspondent BCAAs [65], it is unclear how fast this process is. In fact, we recently demonstrated that an acute increase in BCKA does not lead to a significant change in cardiac BCAA levels. Moreover, we recently developed a mouse model where we deleted BCAT2 in the heart to selectively increase cardiac BCAAs and decrease cardiac BCKAs [48]. The accumulation of cardiac BCAA levels in the Bcat2 knockout hearts did not impact cardiac insulin sensitivity [48]. However, BCAT2 deletion enhances cardiac insulin signalling and insulin-stimulated glucose oxidation rates [48]. These findings demonstrate that it is BCKAs, not BCAAs, that influence cardiac insulin signalling. In further support of this, we recently showed that reducing cardiac BCKA levels by cardiac-specific deletion of Bcat2 mitigates cardiac insulin resistance and enhances insulin-stimulated glucose oxidation rates in the failing heart [66]. This enhancement in cardiac glucose oxidation is mediated, at least in part, via enhancing mitochondrial Akt activity [66]. How BCKAs enhance insulin signalling in the heart remains an interesting scope for future investigations.
Systemic: At the whole-body level, the use of LY3351337 to inhibit both BCAT1 and BCAT2 in Zucker fatty rats results in increased circulating levels of BCAA and glycine [54], with the latter showing an inverse correlation with impaired glucose handling and T2D [67, 68]. This intervention significantly improves glucose tolerance and insulin sensitivity. Similarly, inhibition of BCAT2 with Telmisartan reduces circulating BCKA levels and body weight, leading to notable enhancements in glucose tolerance and insulin sensitivity in mice on a high-fat diet [47]. Furthermore, a plethora of studies utilizing various animal models of obesity and insulin resistance consistently demonstrates that treatment with the BCKDK inhibitor 3,6-dichlorobenzo[b]thiophene-2-carboxylic acid, commonly referred to as BT2, enhances BCAA oxidation, reduces circulating BCAA and BCKA levels, and notably improves glucose tolerance and insulin sensitivity [27, 44, 45, 55]. In a recent randomized and controlled clinical trial, T2D patients receiving sodium phenylbutyrate (4.8g per day), an FDA-approved drug for treating acute hyperammonemia that inhibits BCKDK and promotes BCAA oxidation, exhibited enhanced insulin sensitivity after just 2 weeks of treatment compared to the placebo group [69]. It is worth mentioning that enhancing systemic BCAA oxidation through global deletion of BCKDK in lean mice results in decreased circulating BCAA and BCKA levels [27]. However, this manipulation does not significantly impact glucose disposal in BCKDK knockout lean mice following oral glucose administration. Alternatively, suppressing whole-body BCAA oxidation in mice by deleting PP2Cm increases circulating BCAA and BCKA levels and enhances glucose tolerance and insulin sensitivity, regardless of the presence or absence of obesity and insulin resistance [52, 53].
One pivotal question emerges from these studies: Is it better to augment or reduce BCAA oxidation in obesity and insulin resistance? We argue that enhancing BCAA oxidation enhancing and mainly lowering BCAA and BCKA levels appears to be a more advantageous approach, given the adverse effects associated with suppressing BCAA oxidation and increasing BCAA and BCKA levels, such as ischemia-reperfusion injury and heart failure [49, 50]. Consequently, another fundamental question remains unanswered: Does systemic enhancement of BCAA oxidation alone alleviate insulin resistance, or is it the reduction in BCKAs that alleviates insulin resistance? We propose the latter based on the following evidence: 1) While BT2 administration in animals dephosphorylates and activates the BCKDH complex in multiple organs, resulting in a systemic reduction in plasma BCAA and BCKA levels [45, 70], the magnitude of BCKA reduction appears more pronounced compared to BCAA reduction in ob/ob mice treated with BT2 over a period of 4–6 weeks [55]. 2) Screening efforts aimed at discovering more potent BCKDK inhibitors led to the identification of thiophene PF-07208254 as an allosteric BCKDK inhibitor exhibiting superior potency to BT2 [56]. Both PF-07208254 and BT2 dephosphorylate BCKDH at the same site, resulting in diminished levels of BCKAs and improved glucose tolerance and insulin sensitivity. Intriguingly, structure-activity relationship studies have revealed thiazoles as BCKDK inhibitors with even greater potency than PF-07208254 and BT2. However, despite their ability to dephosphorylate BCKDH, thiazole inhibitors elevate BCKA levels and counteract the favorable effects of PF-07208254 and BT2 by increasing the proximity of BCKDK to BCKDH-E2. 3) In individuals with maple syrup urine disease, the oxidation of BCAAs is hindered due to a deficiency in BCKDH enzyme. It’s noteworthy that despite elevated plasma BCAA levels, these individuals do not typically experience insulin resistance [71–74].
Together, these observations further suggest that primarily reducing systemic levels of BCKAs may enhance insulin sensitivity. However, further research in this area is needed to thoroughly investigate and confirm this hypothesis.
The impact of dietary BCAA supplementation or restriction on insulin resistance
Numerous preclinical studies have indicated that supplementing with BCAAs worsens insulin resistance, while restricting their intake improves insulin sensitivity in various obese animal models (Table 2). Recent evidence further suggests that limiting dietary BCAAs could potentially improve health and longevity in male mice [83], whereas high BCAA consumption induces obesity and shortens lifespan in mice [84]. While many of these studies have treated all three BCAAs as having equivalent metabolic effects, emerging research indicates that each BCAA may exert unique influences on obesity and insulin sensitivity. For instance, Yu and colleagues demonstrated that restricting either isoleucine or valine, but not leucine, enhances glucose tolerance and hepatic insulin sensitivity in mice on a Western diet [81]. Intriguingly, reintroducing either isoleucine or all three BCAAs, but not leucine or valine alone, reverses these metabolic benefits. In another study, the same researchers found that lifelong isoleucine restriction increases lifespan and improves glucose homeostasis in both male and female mice [82]. Similarly, another group observed that valine supplementation in mice on a high-fat diet significantly impairs glucose tolerance and insulin sensitivity [77]. Likewise, a plethora of studies have illustrated that leucine supplementation yields various beneficial effects on glucose homeostasis across different mouse models of obesity and insulin resistance [85–91]. Notably, it is well-documented that leucine increases hypothalamic mTOR signalling while reducing food intake and body weight [92]. Collectively, these findings underscore that each of the individual BCAAs exerts distinct metabolic effects on obesity and insulin sensitivity. Furthermore, accumulating evidence suggests that the elevation of BCAA levels per se may not be the primary driver of insulin resistance [47, 48, 51], but rather their downstream metabolites (such as BCKAs, 3-HIB, and specific acylcarnitine species) that play a pivotal role in triggering the disease. Since each BCAA follows a distinct metabolic pathway after oxidation (Figure 1), this presents promising opportunities to selectively target either the isoleucine, valine, or both pathways to treat and prevent obesity and insulin resistance.
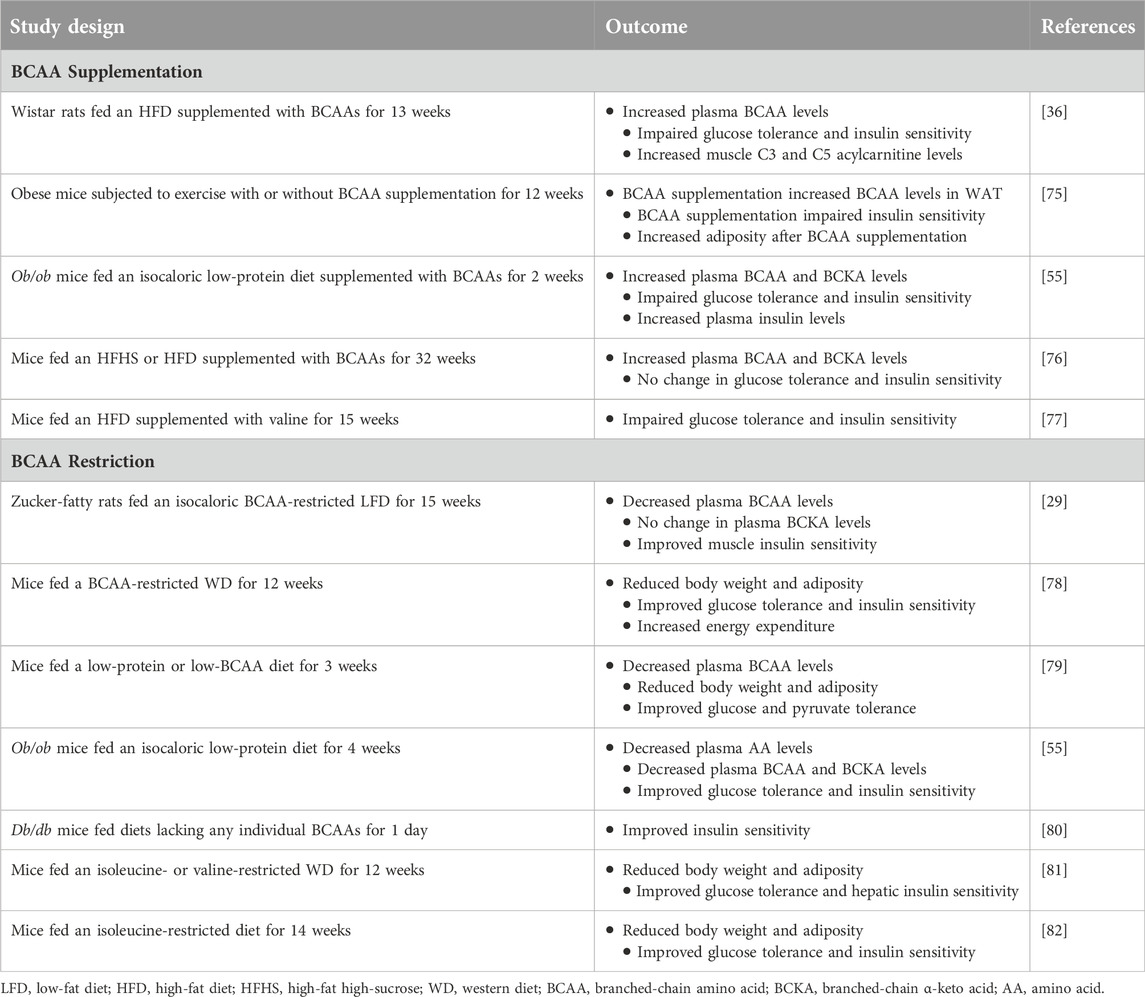
Table 2. Preclinical studies demonstrating the impact of dietary BCAA supplementation or restriction on insulin sensitivity.
Discussion
While elevated plasma levels of BCAAs have consistently been linked to insulin resistance and T2D, recent evidence suggests that the direct implication of BCAAs themselves in insulin resistance may not be significant. Instead, emerging evidence suggests that the accumulation of their downstream metabolites, such as BCKAs, could play a crucial role in exacerbating insulin resistance. If elevated BCKA levels are indeed the main driver of insulin resistance, then lowering them can be accomplished through BCAT2 inhibition or BCKDK inhibition (Figure 2). Further research is needed to determine whether targeting these downstream metabolites of BCAAs could offer a promising avenue for treating and preventing obesity-induced insulin resistance and T2D.
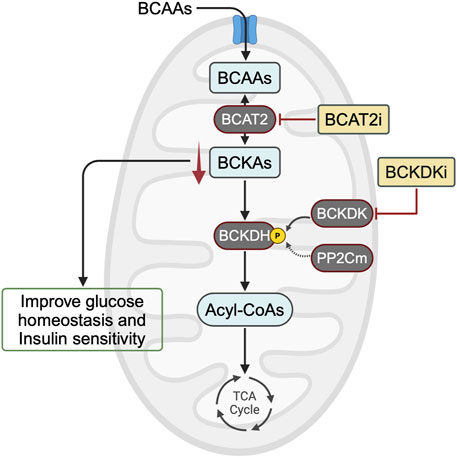
Figure 2. Potential targets for correcting alterations in BCAA metabolism to treat insulin resistance. In animal models of obesity and insulin resistance, mounting evidence suggests that BCKA accumulation, rather than BCAA accumulation, impairs glucose homeostasis and decreases insulin sensitivity. Therefore, one approach to lowering BCKA levels would be to inhibit BCAT2 or enhance BCAA oxidation activity using a BCKDK inhibitor. BCAA, branched-chain amino acid; BCKA, branched-chain α-keto acid; BCAT2, branched-chain amino acid aminotransferase 2; BCKDH, branched-chain α-ketoacid dehydrogenase complex; BCKDK, BCKDH kinase; PP2Cm, protein phosphatase 2Cm; BCAT2i, BCAT2 inhibitor; BCKDKi, BCKDK inhibitor.
Author contributions
RA and AA drafted the initial version of the manuscript. AA created the figures and tables. QK and GL revised the manuscript and contributed to the heart section. All authors contributed to the article and approved the submitted version.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This review was supported by a Seeding Grant from Diabetes Québec to RA. RA is a Research Scholar of the Fonds de Recherche du Québec - Santé (FRQS) and a New Investigator of the Kidney Research Scientist Core Education and National Training (KRESCENT).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Figures 1, 2 was generated using BioRender.com.
References
1. Wang, D, Ye, J, Shi, R, Zhao, B, Liu, Z, Lin, W, et al. Dietary protein and amino acid restriction: roles in metabolic health and aging-related diseases. Free Radic Biol Med (2022) 178:226–42. doi:10.1016/j.freeradbiomed.2021.12.009
2. Ridaura, VK, Faith, JJ, Rey, FE, Cheng, J, Duncan, AE, Kau, AL, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science (2013) 341(6150):1241214. doi:10.1126/science.1241214
3. Pedersen, HK, Gudmundsdottir, V, Nielsen, HB, Hyotylainen, T, Nielsen, T, Jensen, BAH, et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature (2016) 535(7612):376–81. doi:10.1038/nature18646
4. Wolfson, RL, Chantranupong, L, Saxton, RA, Shen, K, Scaria, SM, Cantor, JR, et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science (2016) 351(6268):43–8. doi:10.1126/science.aab2674
5. Caballero, B, Finer, N, and Wurtman, RJ. Plasma amino acids and insulin levels in obesity: response to carbohydrate intake and tryptophan supplements. Metabolism (1988) 37(7):672–6. doi:10.1016/0026-0495(88)90089-3
6. Bagdade, JD, Bierman, EL, and Porte, D. Are plasma amino acid levels elevated in obesity? N Engl J Med (1970) 282(3):166. doi:10.1056/NEJM197001152820316
7. Felig, P, Marliss, E, and Cahill, GF. Plasma amino acid levels and insulin secretion in obesity. N Engl J Med (1969) 281(15):811–6. doi:10.1056/nejm196910092811503
8. Marchesini, G, Bianchi, G, Rossi, B, Muggeo, M, and Bonora, E. Effects of hyperglycaemia and hyperinsulinaemia on plasma amino acid levels in obese subjects with normal glucose tolerance. Int J Obes (2000) 24(5):552–8. doi:10.1038/sj.ijo.0801195
9. Wijekoon, EP, Skinner, C, Brosnan, ME, and Brosnan, JT. Amino acid metabolism in the Zucker diabetic fatty rat: effects of insulin resistance and of type 2 diabetes. Can J Physiol Pharmacol (2004) 82(7):506–14. doi:10.1139/y04-067
10. Shah, SH, Bain, JR, Muehlbauer, MJ, Stevens, RD, Crosslin, DR, Haynes, C, et al. Association of a peripheral blood metabolic profile with coronary artery disease and risk of subsequent cardiovascular events. Circ Cardiovasc Genet (2010) 3(2):207–14. doi:10.1161/circgenetics.109.852814
11. Wang, TJ, Larson, MG, Vasan, RS, Cheng, S, Rhee, EP, McCabe, E, et al. Metabolite profiles and the risk of developing diabetes. Nat Med (2011) 17(4):448–53. doi:10.1038/nm.2307
12. Laferrere, B, Reilly, D, Arias, S, Swerdlow, N, Gorroochurn, P, Bawa, B, et al. Differential metabolic impact of gastric bypass surgery versus dietary intervention in obese diabetic subjects despite identical weight loss. Sci Transl Med (2011) 3(80):80re2. doi:10.1126/scitranslmed.3002043
13. Jensen, MD, and Haymond, MW. Protein metabolism in obesity: effects of body fat distribution and hyperinsulinemia on leucine turnover. Am J Clin Nutr (1991) 53(1):172–6. doi:10.1093/ajcn/53.1.172
14. Luzi, L, Castellino, P, and DeFronzo, RA. Insulin and hyperaminoacidemia regulate by a different mechanism leucine turnover and oxidation in obesity. Am J Physiology-Endocrinology Metab (1996) 270(2 Pt 1):E273–81. doi:10.1152/ajpendo.1996.270.2.e273
15. Harper, AE, Miller, RH, and Block, KP. Branched-chain amino acid metabolism. Annu Rev Nutr (1984) 4:409–54. doi:10.1146/annurev.nutr.4.1.409
16. Blair, MC, Neinast, MD, and Arany, Z. Whole-body metabolic fate of branched-chain amino acids. Biochem J (2021) 478(4):765–76. doi:10.1042/bcj20200686
17. Neinast, M, Murashige, D, and Arany, Z. Branched chain amino acids. Annu Rev Physiol (2019) 81:139–64. doi:10.1146/annurev-physiol-020518-114455
18. Sweatt, AJ, Wood, M, Suryawan, A, Wallin, R, Willingham, MC, and Hutson, SM. Branched-chain amino acid catabolism: unique segregation of pathway enzymes in organ systems and peripheral nerves. Am J Physiology-Endocrinology Metab (2004) 286(1):E64–76. doi:10.1152/ajpendo.00276.2003
19. Suryawan, A, Hawes, JW, Harris, RA, Shimomura, Y, Jenkins, AE, and Hutson, SM. A molecular model of human branched-chain amino acid metabolism. Am J Clin Nutr (1998) 68(1):72–81. doi:10.1093/ajcn/68.1.72
20. Lin, HM, Kaneshige, M, Zhao, L, Zhang, X, Hanover, JA, and Cheng, SY. An isoform of branched-chain aminotransferase is a novel co-repressor for thyroid hormone nuclear receptors. J Biol Chem (2001) 276(51):48196–205. doi:10.1074/jbc.m104320200
21. Hutson, SM, Wallin, R, and Hall, TR. Identification of mitochondrial branched chain aminotransferase and its isoforms in rat tissues. J Biol Chem (1992) 267(22):15681–6. doi:10.1016/s0021-9258(19)49589-6
22. Ruderman, NB, and Berger, M. The formation of glutamine and alanine in skeletal muscle. J Biol Chem (1974) 249(17):5500–6. doi:10.1016/s0021-9258(20)79756-5
23. Odessey, R, Khairallah, EA, and Goldberg, AL. Origin and possible significance of alanine production by skeletal muscle. J Biol Chem (1974) 249(23):7623–9. doi:10.1016/s0021-9258(19)81283-8
24. Lu, G, Ren, S, Korge, P, Choi, J, Dong, Y, Weiss, J, et al. A novel mitochondrial matrix serine/threonine protein phosphatase regulates the mitochondria permeability transition pore and is essential for cellular survival and development. Genes Dev (2007) 21(7):784–96. doi:10.1101/gad.1499107
25. Kainulainen, H, Hulmi, JJ, and Kujala, UM. Potential role of branched-chain amino acid catabolism in regulating fat oxidation. Exerc Sport Sci Rev (2013) 41(4):194–200. doi:10.1097/jes.0b013e3182a4e6b6
26. Brosnan, JT, and Brosnan, ME. Branched-chain amino acids: enzyme and substrate regulation. J Nutr (2006) 136(1 Suppl. l):207s–11s. doi:10.1093/jn/136.1.207s
27. Neinast, MD, Jang, C, Hui, S, Murashige, DS, Chu, Q, Morscher, RJ, et al. Quantitative analysis of the whole-body metabolic fate of branched-chain amino acids. Cel Metab (2019) 29(2):417–29.e4. doi:10.1016/j.cmet.2018.10.013
28. Yoneshiro, T, Wang, Q, Tajima, K, Matsushita, M, Maki, H, Igarashi, K, et al. BCAA catabolism in brown fat controls energy homeostasis through SLC25A44. Nature (2019) 572(7771):614–9. doi:10.1038/s41586-019-1503-x
29. White, PJ, Lapworth, AL, An, J, Wang, L, McGarrah, RW, Stevens, RD, et al. Branched-chain amino acid restriction in Zucker-fatty rats improves muscle insulin sensitivity by enhancing efficiency of fatty acid oxidation and acyl-glycine export. Mol Metab (2016) 5(7):538–51. doi:10.1016/j.molmet.2016.04.006
30. She, P, Van Horn, C, Reid, T, Hutson, SM, Cooney, RN, and Lynch, CJ. Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am J Physiology-Endocrinology Metab (2007) 293(6):E1552–63. doi:10.1152/ajpendo.00134.2007
31. Lian, K, Du, C, Liu, Y, Zhu, D, Yan, W, Zhang, H, et al. Impaired adiponectin signaling contributes to disturbed catabolism of branched-chain amino acids in diabetic mice. Diabetes (2015) 64(1):49–59. doi:10.2337/db14-0312
32. Herman, MA, She, P, Peroni, OD, Lynch, CJ, and Kahn, BB. Adipose tissue branched chain amino acid (BCAA) metabolism modulates circulating BCAA levels. J Biol Chem (2010) 285(15):11348–56. doi:10.1074/jbc.m109.075184
33. Lackey, DE, Lynch, CJ, Olson, KC, Mostaedi, R, Ali, M, Smith, WH, et al. Regulation of adipose branched-chain amino acid catabolism enzyme expression and cross-adipose amino acid flux in human obesity. Am J Physiology-Endocrinology Metab (2013) 304(11):E1175–87. doi:10.1152/ajpendo.00630.2012
34. Zimmerman, HA, Olson, KC, Chen, G, and Lynch, CJ. Adipose transplant for inborn errors of branched chain amino acid metabolism in mice. Mol Genet Metab (2013) 109(4):345–53. doi:10.1016/j.ymgme.2013.05.010
35. Jang, C, Oh, SF, Wada, S, Rowe, GC, Liu, L, Chan, MC, et al. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat Med (2016) 22(4):421–6. doi:10.1038/nm.4057
36. Newgard, CB, An, J, Bain, JR, Muehlbauer, MJ, Stevens, RD, Lien, LF, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cel Metab (2009) 9(4):565–6. doi:10.1016/j.cmet.2009.05.001
37. Newgard, CB. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cel Metab (2012) 15(5):606–14. doi:10.1016/j.cmet.2012.01.024
38. Biswas, D, Dao, KT, Mercer, A, Cowie, AM, Duffley, L, El Hiani, Y, et al. Branched-chain ketoacid overload inhibits insulin action in the muscle. J Biol Chem (2020) 295(46):15597–621. doi:10.1074/jbc.ra120.013121
39. Um, SH, D'Alessio, D, and Thomas, G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cel Metab (2006) 3(6):393–402. doi:10.1016/j.cmet.2006.05.003
40. Tremblay, F, Brûlé, S, Hee Um, S, Li, Y, Masuda, K, Roden, M, et al. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc Natl Acad Sci U S A (2007) 104(35):14056–61. doi:10.1073/pnas.0706517104
41. Tremblay, F, Lavigne, C, Jacques, H, and Marette, A. Role of dietary proteins and amino acids in the pathogenesis of insulin resistance. Annu Rev Nutr (2007) 27:293–310. doi:10.1146/annurev.nutr.25.050304.092545
42. Gojda, J, and Cahova, M. Gut microbiota as the link between elevated BCAA serum levels and insulin resistance. Biomolecules (2021) 11(10):1414. doi:10.3390/biom11101414
43. Choi, BS, Daniel, N, Houde, VP, Ouellette, A, Marcotte, B, Varin, TV, et al. Feeding diversified protein sources exacerbates hepatic insulin resistance via increased gut microbial branched-chain fatty acids and mTORC1 signaling in obese mice. Nat Commun (2021) 12(1):3377. doi:10.1038/s41467-021-23782-w
44. Blair, MC, Neinast, MD, Jang, C, Chu, Q, Jung, JW, Axsom, J, et al. Branched-chain amino acid catabolism in muscle affects systemic BCAA levels but not insulin resistance. Nat Metab (2023) 5(4):589–606. doi:10.1038/s42255-023-00794-y
45. White, PJ, McGarrah, RW, Grimsrud, PA, Tso, SC, Yang, WH, Haldeman, JM, et al. The BCKDH kinase and phosphatase integrate BCAA and lipid metabolism via regulation of ATP-citrate lyase. Cel Metab (2018) 27(6):1281–93.e7. doi:10.1016/j.cmet.2018.04.015
46. Ananieva, EA, Van Horn, CG, Jones, MR, and Hutson, SM. Liver BCATm transgenic mouse model reveals the important role of the liver in maintaining BCAA homeostasis. J Nutr Biochem (2017) 40:132–40. doi:10.1016/j.jnutbio.2016.10.014
47. Ma, QX, Zhu, WY, Lu, XC, Jiang, D, Xu, F, Li, JT, et al. BCAA-BCKA axis regulates WAT browning through acetylation of PRDM16. Nat Metab (2022) 4(1):106–22. doi:10.1038/s42255-021-00520-6
48. Uddin, GM, Karwi, QG, Pherwani, S, Gopal, K, Wagg, CS, Biswas, D, et al. Deletion of BCATm increases insulin-stimulated glucose oxidation in the heart. Metabolism (2021) 124:154871. doi:10.1016/j.metabol.2021.154871
49. Li, T, Zhang, Z, Kolwicz, SC, Abell, L, Roe, ND, Kim, M, et al. Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia-reperfusion injury. Cel Metab (2017) 25(2):374–85. doi:10.1016/j.cmet.2016.11.005
50. Sun, H, Olson, KC, Gao, C, Prosdocimo, DA, Zhou, M, Wang, Z, et al. Catabolic defect of branched-chain amino acids promotes heart failure. Circulation (2016) 133(21):2038–49. doi:10.1161/circulationaha.115.020226
51. She, P, Reid, TM, Bronson, SK, Vary, TC, Hajnal, A, Lynch, CJ, et al. Disruption of BCATm in mice leads to increased energy expenditure associated with the activation of a futile protein turnover cycle. Cel Metab (2007) 6(3):181–94. doi:10.1016/j.cmet.2007.08.003
52. Nishi, K, Yoshii, A, Abell, L, Zhou, B, Frausto, R, Ritterhoff, J, et al. Branched-chain keto acids inhibit mitochondrial pyruvate carrier and suppress gluconeogenesis in hepatocytes. Cel Rep (2023) 42(6):112641. doi:10.1016/j.celrep.2023.112641
53. Wang, J, Liu, Y, Lian, K, Shentu, X, Fang, J, Shao, J, et al. BCAA catabolic defect alters glucose metabolism in lean mice. Front Physiol (2019) 10:1140. doi:10.3389/fphys.2019.01140
54. White, PJ, Lapworth, AL, McGarrah, RW, Kwee, LC, Crown, SB, Ilkayeva, O, et al. Muscle-liver trafficking of BCAA-derived nitrogen underlies obesity-related Glycine depletion. Cel Rep (2020) 33(6):108375. doi:10.1016/j.celrep.2020.108375
55. Zhou, M, Shao, J, Wu, CY, Shu, L, Dong, W, Liu, Y, et al. Targeting BCAA catabolism to treat obesity-associated insulin resistance. Diabetes (2019) 68(9):1730–46. doi:10.2337/db18-0927
56. Roth Flach, RJ, Bollinger, E, Reyes, AR, Laforest, B, Kormos, BL, Liu, S, et al. Small molecule branched-chain ketoacid dehydrogenase kinase (BDK) inhibitors with opposing effects on BDK protein levels. Nat Commun (2023) 14(1):4812. doi:10.1038/s41467-023-40536-y
57. White, PJ, McGarrah, RW, Herman, MA, Bain, JR, Shah, SH, and Newgard, CB. Insulin action, type 2 diabetes, and branched-chain amino acids: a two-way street. Mol Metab (2021) 52:101261. doi:10.1016/j.molmet.2021.101261
58. Hsiao, G, Chapman, J, Ofrecio, JM, Wilkes, J, Resnik, JL, Thapar, D, et al. Multi-tissue, selective PPARγ modulation of insulin sensitivity and metabolic pathways in obese rats. Am J Physiology-Endocrinology Metab (2011) 300(1):E164–74. doi:10.1152/ajpendo.00219.2010
59. Adams, SH. Emerging perspectives on essential amino acid metabolism in obesity and the insulin-resistant state. Adv Nutr (2011) 2(6):445–56. doi:10.3945/an.111.000737
60. Green, CR, Wallace, M, Divakaruni, AS, Phillips, SA, Murphy, AN, Ciaraldi, TP, et al. Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat Chem Biol (2016) 12(1):15–21. doi:10.1038/nchembio.1961
61. Wallace, M, Green, CR, Roberts, LS, Lee, YM, McCarville, JL, Sanchez-Gurmaches, J, et al. Enzyme promiscuity drives branched-chain fatty acid synthesis in adipose tissues. Nat Chem Biol (2018) 14(11):1021–31. doi:10.1038/s41589-018-0132-2
62. Karwi, QG, and Lopaschuk, GD. Branched-chain amino acid metabolism in the failing heart. Cardiovasc Drugs Ther (2023) 37(2):413–20. doi:10.1007/s10557-022-07320-4
63. Fillmore, N, Wagg, CS, Zhang, L, Fukushima, A, and Lopaschuk, GD. Cardiac branched-chain amino acid oxidation is reduced during insulin resistance in the heart. Am J Physiology-Endocrinology Metab (2018) 315:E1046–E1052. doi:10.1152/ajpendo.00097.2018
64. Uddin, GM, Zhang, L, Shah, S, Fukushima, A, Wagg, CS, Gopal, K, et al. Impaired branched chain amino acid oxidation contributes to cardiac insulin resistance in heart failure. Cardiovasc Diabetol (2019) 18(1):86. doi:10.1186/s12933-019-0892-3
65. Walejko, JM, Christopher, BA, Crown, SB, Zhang, GF, Pickar-Oliver, A, Yoneshiro, T, et al. Branched-chain α-ketoacids are preferentially reaminated and activate protein synthesis in the heart. Nat Commun (2021) 12(1):1680. doi:10.1038/s41467-021-21962-2
66. Karwi, Q, Uddin, GM, Wagg, CS, and Lopaschuk, GD. Abstract MP125: branched-chain keto acids, not branched-chain amino acids, impairs cardiac insulin sensitivity by disrupting insulin signaling in the mitochondria. Circ Res (2020) 127(Suppl. l_1):AMP125. doi:10.1161/res.127.suppl_1.mp125
67. Wang-Sattler, R, Yu, Z, Herder, C, Messias, AC, Floegel, A, He, Y, et al. Novel biomarkers for pre-diabetes identified by metabolomics. Mol Syst Biol (2012) 8:615. doi:10.1038/msb.2012.43
68. Floegel, A, Stefan, N, Yu, Z, Mühlenbruch, K, Drogan, D, Joost, HG, et al. Identification of serum metabolites associated with risk of type 2 diabetes using a targeted metabolomic approach. Diabetes (2013) 62(2):639–48. doi:10.2337/db12-0495
69. Vanweert, F, Neinast, M, Tapia, EE, van de Weijer, T, Hoeks, J, Schrauwen-Hinderling, VB, et al. A randomized placebo-controlled clinical trial for pharmacological activation of BCAA catabolism in patients with type 2 diabetes. Nat Commun (2022) 13(1):3508. doi:10.1038/s41467-022-31249-9
70. Tso, SC, Gui, WJ, Wu, CY, Chuang, JL, Qi, X, Skvorak, KJ, et al. Benzothiophene carboxylate derivatives as novel allosteric inhibitors of branched-chain α-ketoacid dehydrogenase kinase. J Biol Chem (2014) 289(30):20583–93. doi:10.1074/jbc.m114.569251
71. Schadewaldt, P, Bodner-Leidecker, A, Hammen, HW, and Wendel, U. Significance of L-alloisoleucine in plasma for diagnosis of maple syrup urine disease. Clin Chem (1999) 45(10):1734–40. doi:10.1093/clinchem/45.10.1734
72. Podebrad, F, Heil, M, Reichert, S, Mosandl, A, Sewell, AC, and Böhles, H. 4,5-dimethyl-3-hydroxy-2[5H]-furanone (sotolone)--the odour of maple syrup urine disease. J Inherit Metab Dis (1999) 22(2):107–14. doi:10.1023/a:1005433516026
73. Sewell, AC, Mosandl, A, and Böhles, H. False diagnosis of maple syrup urine disease owing to ingestion of herbal tea. N Engl J Med (1999) 341(10):769. doi:10.1056/nejm199909023411020
74. Gambello, MJ, and Li, H. Current strategies for the treatment of inborn errors of metabolism. J Genet Genomics (2018) 45(2):61–70. doi:10.1016/j.jgg.2018.02.001
75. Zhang, H, Xiang, L, Huo, M, Wu, Y, Yu, M, Lau, CW, et al. Branched-chain amino acid supplementation impairs insulin sensitivity and promotes lipogenesis during exercise in diet-induced obese mice. Obesity (Silver Spring) (2022) 30(6):1205–18. doi:10.1002/oby.23394
76. Lee, J, Vijayakumar, A, White, PJ, Xu, Y, Ilkayeva, O, Lynch, CJ, et al. BCAA supplementation in mice with diet-induced obesity alters the metabolome without impairing glucose homeostasis. Endocrinology (2021) 162(7):bqab062. doi:10.1210/endocr/bqab062
77. Ma, Q, Hu, L, Zhu, J, Chen, J, Wang, Z, Yue, Z, et al. Valine supplementation does not reduce lipid accumulation and improve insulin sensitivity in mice fed high-fat diet. ACS Omega (2020) 5(48):30937–45. doi:10.1021/acsomega.0c03707
78. Cummings, NE, Williams, EM, Kasza, I, Konon, EN, Schaid, MD, Schmidt, BA, et al. Restoration of metabolic health by decreased consumption of branched-chain amino acids. J Physiol (2018) 596(4):623–45. doi:10.1113/jp275075
79. Fontana, L, Cummings, NE, Arriola Apelo, SI, Neuman, JC, Kasza, I, Schmidt, BA, et al. Decreased consumption of branched-chain amino acids improves metabolic health. Cel Rep (2016) 16(2):520–30. doi:10.1016/j.celrep.2016.05.092
80. Xiao, F, Yu, J, Guo, Y, Deng, J, Li, K, Du, Y, et al. Effects of individual branched-chain amino acids deprivation on insulin sensitivity and glucose metabolism in mice. Metabolism (2014) 63(6):841–50. doi:10.1016/j.metabol.2014.03.006
81. Yu, D, Richardson, NE, Green, CL, Spicer, AB, Murphy, ME, Flores, V, et al. The adverse metabolic effects of branched-chain amino acids are mediated by isoleucine and valine. Cel Metab (2021) 33(5):905–22.e6. doi:10.1016/j.cmet.2021.03.025
82. Green, CL, Trautman, ME, Chaiyakul, K, Jain, R, Alam, YH, Babygirija, R, et al. Dietary restriction of isoleucine increases healthspan and lifespan of genetically heterogeneous mice. Cel Metab (2023) 35(11):1976–95.e6. doi:10.1016/j.cmet.2023.10.005
83. Richardson, NE, Konon, EN, Schuster, HS, Mitchell, AT, Boyle, C, Rodgers, AC, et al. Lifelong restriction of dietary branched-chain amino acids has sex-specific benefits for frailty and lifespan in mice. Nat Aging (2021) 1(1):73–86. doi:10.1038/s43587-020-00006-2
84. Solon-Biet, SM, Cogger, VC, Pulpitel, T, Wahl, D, Clark, X, Bagley, EE, et al. Branched chain amino acids impact health and lifespan indirectly via amino acid balance and appetite control. Nat Metab (2019) 1(5):532–45. doi:10.1038/s42255-019-0059-2
85. Zhang, Y, Guo, K, LeBlanc, RE, Loh, D, Schwartz, GJ, and Yu, YH. Increasing dietary leucine intake reduces diet-induced obesity and improves glucose and cholesterol metabolism in mice via multimechanisms. Diabetes (2007) 56(6):1647–54. doi:10.2337/db07-0123
86. Freudenberg, A, Petzke, KJ, and Klaus, S. Comparison of high-protein diets and leucine supplementation in the prevention of metabolic syndrome and related disorders in mice. J Nutr Biochem (2012) 23(11):1524–30. doi:10.1016/j.jnutbio.2011.10.005
87. Li, H, Xu, M, Lee, J, He, C, and Xie, Z. Leucine supplementation increases SIRT1 expression and prevents mitochondrial dysfunction and metabolic disorders in high-fat diet-induced obese mice. Am J Physiology-Endocrinology Metab (2012) 303(10):E1234–44. doi:10.1152/ajpendo.00198.2012
88. Fu, L, Bruckbauer, A, Li, F, Cao, Q, Cui, X, Wu, R, et al. Leucine amplifies the effects of metformin on insulin sensitivity and glycemic control in diet-induced obese mice. Metabolism (2015) 64(7):845–56. doi:10.1016/j.metabol.2015.03.007
89. Guo, K, Yu, YH, Hou, J, and Zhang, Y. Chronic leucine supplementation improves glycemic control in etiologically distinct mouse models of obesity and diabetes mellitus. Nutr Metab (Lond) (2010) 7:57. doi:10.1186/1743-7075-7-57
90. Binder, E, Bermúdez-Silva, FJ, Elie, M, Leste-Lasserre, T, Belluomo, I, Clark, S, et al. Leucine supplementation modulates fuel substrates utilization and glucose metabolism in previously obese mice. Obesity (Silver Spring) (2014) 22(3):713–20. doi:10.1002/oby.20578
91. Nairizi, A, She, P, Vary, TC, and Lynch, CJ. Leucine supplementation of drinking water does not alter susceptibility to diet-induced obesity in mice. J Nutr (2009) 139(4):715–9. doi:10.3945/jn.108.100081
Keywords: BCAAs, BCKAs, obesity, insulin resistance, type 2 diabetes
Citation: Abdualkader AM, Karwi QG, Lopaschuk GD and Al Batran R (2024) The role of branched-chain amino acids and their downstream metabolites in mediating insulin resistance. J. Pharm. Pharm. Sci 27:13040. doi: 10.3389/jpps.2024.13040
Received: 26 March 2024; Accepted: 19 June 2024;
Published: 28 June 2024.
Edited by:
John Reyes Ussher, University of Alberta, CanadaCopyright © 2024 Abdualkader, Karwi, Lopaschuk and Al Batran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rami Al Batran, cmFtaS5hbC5iYXRyYW5AdW1vbnRyZWFsLmNh