 K. Modarage
K. Modarage P. Goggolidou
P. Goggolidou- Department of Biomedical Science and Physiology, Faculty of Science and Engineering, University of Wolverhampton, Wolverhampton, United Kingdom
The definition of a rare disease in the European Union describes genetic disorders that affect less than 1 in 2,000 people per individual disease; collectively these numbers amount to millions of individuals globally, who usually manifest a rare disease early on in life. At present, there are at least 8,000 known rare conditions, of which only some are clearly molecularly defined. Over the recent years, the use of genetic diagnosis is gaining ground into informing clinical practice, particularly in the field of rare diseases, where diagnosis is difficult. To demonstrate the complexity of genetic diagnosis for rare diseases, we focus on Ciliopathies as an example of a group of rare diseases where an accurate diagnosis has proven a challenge and novel practices driven by scientists are needed to help bridge the gap between clinical and molecular diagnosis. Current diagnostic difficulties lie with the vast multitude of genes associated with Ciliopathies and trouble in distinguishing between Ciliopathies presenting with similar phenotypes. Moreover, Ciliopathies such as Autosomal Recessive Polycystic Kidney Disease (ARPKD) and Meckel-Gruber syndrome (MKS) present with early phenotypes and may require the analysis of samples from foetuses with a suspected Ciliopathy. Advancements in Next Generation Sequencing (NGS) have now enabled assessing a larger number of target genes, to ensure an accurate diagnosis. The aim of this review is to provide an overview of current diagnostic techniques relevant to Ciliopathies and discuss the applications and limitations associated with these techniques.
Introduction
Ciliopathies describe a group of disorders that arise due to mutations in cilia, resulting in their abnormal formation or function1. The term entails a group of around 35 reported disorders, with effects seen in multiple organs1. Cilia are short extracellular structures, projecting from the cell membrane that can be classified into motile or immotile (also known as primary) cilia1. Both motile and primary cilia begin to form during the G0/G1 phase of the cell cycle2,3. Motile cilia comprise of nine pairs of microtubules surrounding an additional central pair (9 + 2 arrangement) and present as multiple cilia per cell (Figure 1)2,3. They are usually longer than primary cilia and consist of specialised ciliary motor and accessory proteins, allowing the structure to actively bend and undergo coordinated beating patterns that create flow over the cell surface4. Primary cilia consist of microtubules arranged in a circular pattern, as nine microtubule doublets (9 + 0 arrangement)5; they play an essential mechanosensory role and have been implicated in various cell signalling pathways. One exception is the nodal cilium, a structure which presents with a 9 + 0 arrangement, however, displays directional movement6. In this review, the structure of the cilium will be broadly classified and discussed as two separate compartments – the base of the cilium and intraflagellar transport (IFT).
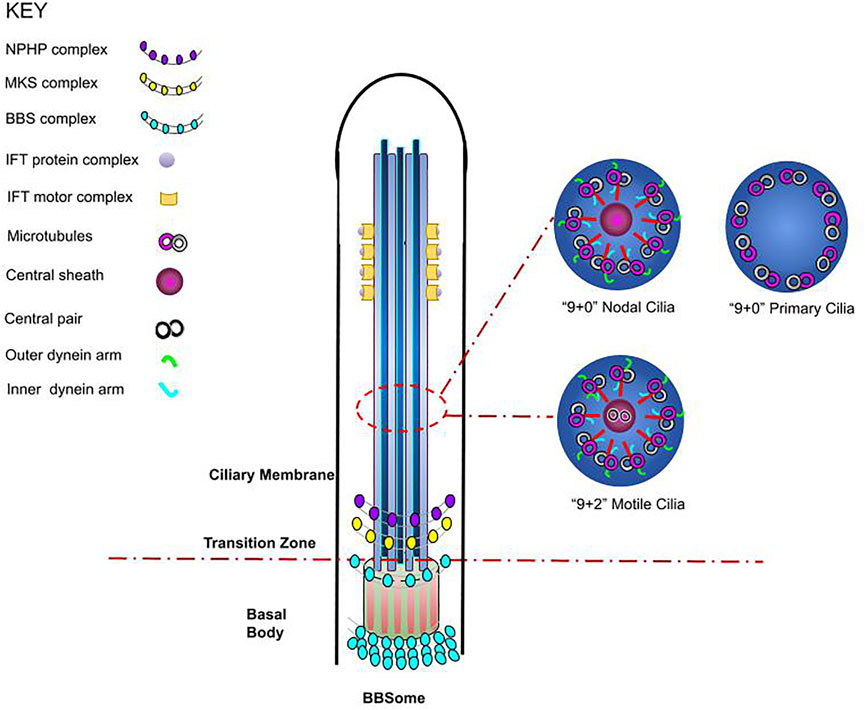
FIGURE 1. Schematic representation of cilium structure. The cilium is a membrane-bounded sensory organelle that is composed of the ciliary membrane, axoneme, and basal body. The ciliary membrane and axoneme form the upper region of the cilium, whereby nine peripheral microtubule doublets are present, whilst in the junction on the basal body, the transition fibres and key ciliary complexes are found3,5. In addition, a cross section of cilia axoneme illustrates the microtubule arrangements in nodal, motile, and non-motile (primary) cilia, highlighting the differences in cilium structure3,5.
The basal body lies at the base of the cilium, where the ciliary gate is located (Figure 1). The ciliary gate is composed of three main structures: the transition fibres (TFs), the transition zone (TZ) and the ciliary necklace and it behaves as a docking site for IFT particles1,5,6,7. The gate is required for ciliogenesis by functioning as a diffusion barrier speculated to aid the transport of proteins and assembled IFT machinery through the formation of pore-like structures1,5,7,8; multiple TZ proteins have been identified to belong to either the Nephronophthisis (NPHP) or the Meckel Gruber Syndrome (MKS) complexes9. IFT has also been associated with the BBSome, where either independently or through cooperation with the BBSome, it is essential for the formation of the ciliary membrane (Figure 1)1,10. The BBSome is a complex of Bardet-Biedl syndrome (BBS) proteins, and it is yet to be established whether this structure is an additional constituent carried by IFT or an integral component of IFT machinery11,12. The correct formation of the above ciliary structural components is essential for their function, with mutations and defects in ciliary genes resulting in the onset of several Ciliopathies such as NPHP, MKS and BBS1,5,7. Since only a few disorders have a known molecular mechanism and there is overlap in the manifestation of many Ciliopathies, challenges are commonly faced when diagnosing these diseases.
Historic and Current Diagnostic Approaches for Ciliopathies
Linkage analysis uses microsatellite markers for the analysis of genes under investigation and it has historically been a commonly practiced technique for the diagnosis of Polycystic Kidney Disease (PKD)13. In Autosomal Dominant Polycystic Kidney Disease (ADPKD), symptoms manifest between the ages of 30–501,14,15 and include renal cysts, hypertension, gross haematuria, nephrolithiasis and urinary tract infections1,14,15. Extra-renal manifestations involve the development of hepatic, pancreatic and thyroid cysts and intracranial aneurysms1,14,15. Comparatively, ARPKD is common in neonates and infants1,14 and may manifest by bilateral enlargement of kidneys, the formation of fluid-filled cysts throughout the collecting ducts of the kidney, oligohydramnios, pulmonary hypoplasia, hepatic fibrosis, and respiratory insufficiency1,14,15. Defective genes associated with ADPKD include Polycystic Kidney Disease 1/2 (PKD1/2), with PKD1 accounting for most ADPKD cases14 (Table 1). In comparison, ARPKD has been associated with mutations in Polycystic Kidney and Hepatic Disease 1 (PKHD1), with recent findings linking mutations in DAZ interacting protein 1-like (DZIP1L) with moderate ARPKD17. PKD1/2 and PKHD1 encode for Polycystin (PC)-1/2 and Fibrocystin respectively, proteins that localise to the cilium1,4,5,14,15.

TABLE 1. A summary of selected Ciliopathies, including their associated genes and their current and recommended molecular diagnostic techniques.
Although linkage analysis helps identify markers that co-segregate with genes of interest, whilst also allowing for patient diagnosis confirmation in cases where mutation positions remain unknown16 and it can be effectively used for neonatal/prenatal testing and can identify the carrier status of at-risk females17, it also has limitations. Linkage analysis has got the risk of recombination, which could result in incorrect carrier status and the rise of de novo mutations/mosaicisms16 and it fails to provide details on the exact mutation type17,18,19. Another major difficulty associated with this technique is the requirement for multiple family members or generations for informative linkage analysis16,19, hence why direct mutation screening has become a more commonly practiced method of diagnosis.
Direct mutation screening is one of the most common and cost-effective methods used to diagnose Ciliopathies such as ADPKD and ARPKD, where the causative genes are known. The technique itself involves sequencing exonic regions of a particular gene, whilst flanking intronic regions, providing details about the mutation position and type19. Positively, samples for direct mutation screening are often easy to obtain with results being relatively definitive16,19,20. Furthermore, screening is now offered during neonatal and prenatal periods, making the technique more applicable to patients. In ARPKD, a definitive prenatal diagnosis can be carried out via direct mutation screening for disease-causing alleles after amniocentesis and chorionic villus sampling21. Unfortunately, this method of testing is only routinely offered to ‘at risk’ families, whilst the invasive nature of these methods also carries a risk of miscarriage at an approximate rate of 0.5–1%21.
It should be noted that direct mutation screening analysis can be useful in identifying isolated probands or in cases where de novo mutations arise but it can become quite restricting in cases where there are rarer mutations present or defects in other genes that are causative of the presenting phenotype16. For example, despite current knowledge regarding ADPKD, genes such as HNF-1β; neutral α-glucosidase AB (GANAB) and DnaJ Heat Shock Protein Family (Hsp40) Member B11 (DNAJB11) have recently been associated with presenting ADPKD-like phenotypes22,23,24. Similarly, mutations in DZIP1L have recently been associated with ARPKD with current efforts being made to identify other modifier genes in ARPKD25. Moreover, some phenotypes of cystic kidney disorders e.g., ADPKD, ARPKD, NPHP and HNF1β-related disease may overlap (e.g. cystic expansion of the kidneys, kidney enlargement, liver fibrosis, situs inversus), making the identification of causative genes and mutations a complex and expensive process. Importantly, it can become complicated to sequence genes such as PKD1 due to the presence of pseudogenes, which have a sequence homology of around 97.6–97.8%, making it challenging for the sensitivity of current diagnostic procedures to identify genuine PKD1 regions26. In addition, in MKS, a highly heterogenous autosomal recessive ciliopathy that is lethal in utero or immediately after birth 1,14,27 and manifests a broad, multi-organ phenotype with considerable variations, key hallmarks of which include pulmonary hypoplasia and cystic kidney dysplasia 1,4,5,14,27, foetal sonographic abnormalities are detected in only ∼2.5% of pregnancies28, meaning that many cases will not be referred for direct mutation screening. Other techniques are also employed to detect known mutations, including Next Generation Sequencing (NGS) and quantitative PCR (qPCR) also known as, Real-time PCR (RT-PCR)29. When RT-PCR is selected as the molecular diagnostic technique in Preimplantation Genetic Diagnosis (PGD), TaqMan, which uses a labelled oligonucleotide (probe) with a fluorophore and a quencher probe at the 5′ and 3′ end respectively, is the most used assay29,30. The limited quantity of material available poses a great technical challenge in PGD, especially in cases of diagnosing heterogeneous disorders like MKS and in certain cases, fresh embryo transfer is necessary, typically 24 h after embryo biopsy to obtain a rapid genotype result31.
“Evolved” PCR as a Method To Accurately Diagnose Ciliopathies
Advanced molecular diagnostic techniques in their entirety have evolved from the Sanger method, the “first-generation” DNA sequencing technique. Nowadays, in clinical practice, the Sanger method is used to validate NGS data, however, this additional level of assurance is not without fault, as it is costly, time-consuming, and not error-free31.
Multiplex qPCR expands on the advantages of RT-PCR, and it is a probe-based assay, where each probe is labelled with a unique fluorescent dye, allowing for simultaneous and rapid amplification of multiple genes in a single reaction32. Multiplex PCR is an ideal diagnostic technique for complicated heterogeneous Ciliopathies like ADPKD. ADPKD shows extensive allelic heterogeneity with six highly homologous sequences of PKD1 exons 1–32, making molecular diagnosis complicated26. Multiplex qPCR was indispensable for detecting mutations in ADPKD patients that were overlooked by NGS33. In a study of 111 ADPKD patients, 86.6% of mutations were detected by NGS, however, one point mutation in exon 1 of PKD1 and five deletions were overlooked by NGS and were only detected using multiplex qPCR33. For the six patients whose mutations went undetected, without multiplex qPCR, it is highly probable that they would have been provided a false negative result that could have led to delay in treatment33.
Another ciliopathy that exhibits similar molecular diagnostic complications to ADPKD is NPHP. NPHP is an autosomal recessive disease that leads to progressive renal failure and manifests as reduced kidney size, loss of corticomedullary differentiation and corticomedullary cysts, together with polyuria, polydipsia, anaemia, growth retardation and hypertension1,14,34. It relies on an initial clinical diagnosis via ultrasonography, followed by renal biopsy analysis, depending on the type of NPHP34. More than 25 genes have been associated with NPHP, with multiple NPHP genes being implicated with the onset of Joubert Syndrome and MKS34. In cases, where the most frequent mutation (NPHP1) is not identified as the pathogenic variant, Long-Read (LR) technologies that can detect repetitive regions of the genome i.e., gene conversion events and are able to generate accurate assemblies35 are the preferred diagnostic method. Additionally, LR technologies can span both the low complexity and repetitive regions, meaning that they can detect paralogue regions of the genome, families of genes, and pseudogene homologs that are often overlooked by short read (SR) technologies35. It has been shown that LR PCR was successful in providing a more reliable ADPKD diagnostic result, when it targeted exons 1–32 of PKD1, generating amplicons from these regions and omitting pseudo-regions26.
The benefits of utilising LR technologies for not only diagnosing ciliopathies but other disorders outweigh the costs, which is often the main limitation associated with this technique. Nevertheless, over the past recent years, the cost has significantly declined, indicating that in the foreseeable future, more LR technologies will be utilised in clinical practice35,36.
The Advances in Sequencing Technologies for Greater and More Accurate Data Capture in Ciliopathies
Targeted Exome Sequencing (TES), which concentrates on a specific panel of genes associated with disease pathogenesis and offers greater sequencing depth, reduced costs, and reduced data burden37, can comprehensively screen heterozygous carriers for a panel of known BBS genes, i.e., 17 causative genes with a total of 242 coding fragments38 to identify pleiotropic disorders that have mutations in large genomic regions like in BBS.
Another option to advance current diagnostic practice for rare diseases is to implement Whole Exome Sequencing (WES). WES involves the sequencing of whole exomes in a genome and has improved sensitivity and efficiency, together with reduced costs37. 1–2% of DNA is comprised of exons, meaning that WES can analyse over 85% of all disease-causing mutations39. Implementing prenatal WES in suspected cases of MKS can help identify pathogenic variants39. In NPHP, a study has highlighted that using a combined approach of multiplex PCR and WES could help overcome the challenges of selecting the best candidate gene based on the phenotypic presentation of the disease40. Overall, WES could lead to an earlier and definitive diagnosis, aiding informed genetic counselling.
In some instances, Whole Genome Sequencing (WGS) which involves sequencing an individual’s entire genome, including coding and non-coding regions41 could be a more appropriate diagnostic method. For genetically heterogeneous disorders such as MKS, BBS, NPHP, the ability to concurrently sequence multiple disease-causing genes using WGS is crucial41,42,43. In ADPKD, WGS is capable of overcoming pseudogene homology complications, whilst in neonates with suspected ARPKD, STATseq, a method of WGS, can complete relevant data analysis within 26–50 h, allowing for rapid confirmation of disease42. Overall, the use of WGS dramatically eases pressure on clinicians and patients, whilst providing the potential for early diagnosis and aiding informative genetic counselling.
Using WGS is a straight-forward process that avoids the time-consuming procedure of targeted sequencing, allowing clinical professionals to identify all variant types in a single test41,42. This includes the detection of single-nucleotide variants, indels, large copy number variants as well as structural variants such as inversions and translocations41. In one ADPKD study, disease-causing variants were identified in 86% of patients using WGS, highlighting the effectiveness and informative potential of WGS as a diagnostic procedure41,42. Unfortunately, WGS has a relatively high cost, making it a more difficult diagnostic tool to access. However, recent advancements in the field, such as the use of Illumina HiSeq X sequencing system has reduced WGS costs41. In 2012, the 100,000 Genomes Project was launched in a bid to advance molecular diagnostics in the United Kingdom and enhance existing knowledge and research into cancer and rare disease44. The project is one of the first national health care systems that routinely uses WGS to sequence and identify any variations in around 85,000 National Health Service (NHS) patients, providing insight on disease aetiology, whilst advancing diagnostic and therapeutic prospects44.
Conclusion
Attempting to identify the pathogenic variants within Ciliopathies, it is clear to see why advancements in molecular diagnostic techniques are undeniably needed. Current limitations lie with the time-consuming manner of diagnostic techniques due to the heterogeneity of Ciliopathies. The overlapping phenotypic presentations together with unclear molecular diagnostics in certain Ciliopathies pose limitations to techniques like direct mutation screening and linkage analysis. Although WES is more expensive than conventional Sanger sequencing, in the broader picture it is a more cost-effective tool. By employing WES as early as possible for diagnostic purposes, the financial burden on health services can be reduced, since multiple tests are not required to confirm the diagnosis, which can often also delay the start of treatment. Furthermore, disease management decisions can be based on a greater depth of information, increasing the reliability of the undertaken approach and informing accurate genetic counselling. Since WES has the capability of screening up to around 85% of a gene, it has a greater likelihood of identifying disease causing mutations than the currently employed techniques. Nevertheless, in diseases such as ADPKD, where pseudogene homology may pose complications in accurate genetic diagnosis, WGS would be the technique of choice. However, one of the major drawbacks of WGS is the cost and thus our suggestion is that WES became a commonly employed gold-standard technique in the field of biomedical science and diagnosis before further advancements to WGS.
Author Contributions
KM, SM, and PG all together devised the idea for the review, wrote the manuscript and prepared the figure and table.
Funding
KM has received funding from the Institute of Biomedical Science. PG is funded by the PKD charity United Kingdom (grant ref: S<ARPKD-TR>).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Reiter, JF, and Leroux, MR. Genes and Molecular Pathways Underpinning Ciliopathies. Nat Rev Mol Cel Biol (2017) 18(9):533–47. doi:10.1038/nrm.2017.60
2. Tucker, RW, Pardee, AB, and Fujiwara, K. Centriole Ciliation Is Related to Quiescence and DNA Synthesis in 3T3 Cells. Cell (1979) 17(3):527–35. doi:10.1016/0092-8674(79)90261-7
3. Tucker, RW, Scher, CD, and Stiles, CD. Centriole Deciliation Associated with the Early Response of 3T3 Cells to Growth Factors but Not to SV40. Cell (1979) 18(4):1065–72. doi:10.1016/0092-8674(79)90219-8
4. Ford, MJ, Yeyati, PL, Mali, GR, Keighren, MA, Waddell, SH, Mjoseng, HK, et al. A Cell/cilia Cycle Biosensor for Single-Cell Kinetics Reveals Persistence of Cilia after G1/S Transition Is a General Property in Cells and Mice. Dev Cel (2018) 47(4):509–23. doi:10.1016/j.devcel.2018.10.027
5. Pala, R, Alomari, N, and Nauli, S. Primary Cilium-dependent Signaling Mechanisms. Ijms (2017) 18(11):2272. doi:10.3390/ijms18112272
6. Buceta, J, Ibañes, M, Rasskin-Gutman, D, Okada, Y, Hirokawa, N, and Izpisúa-Belmonte, JC. Nodal Cilia Dynamics and the Specification of the Left/right axis in Early Vertebrate Embryo Development. Biophysical J (2005) 89(4):2199–209. doi:10.1529/biophysj.105.063743
7. Praveen, K, Davis, EE, and Katsanis, N. Unique Among Ciliopathies: Primary Ciliary Dyskinesia, a Motile Cilia Disorder. F1000prime Rep (2015) 7:36. doi:10.12703/P7-36
8. Milenkovic, L, Scott, MP, and Rohatgi, R. Lateral Transport of Smoothened from the Plasma Membrane to the Membrane of the Cilium. J Cel Biol (2009) 187(3):365–74. doi:10.1083/jcb.200907126
9. Garcia-Gonzalo, FR, Corbit, KC, Sirerol-Piquer, MS, Ramaswami, G, Otto, EA, Noriega, TR, et al. A Transition Zone Complex Regulates Mammalian Ciliogenesis and Ciliary Membrane Composition. Nat Genet (2011) 43(8):776–84. doi:10.1038/ng.891
10. Jin, H, White, SR, Shida, T, Schulz, S, Aguiar, M, Gygi, SP, et al. The Conserved Bardet-Biedl Syndrome Proteins Assemble a Coat that Traffics Membrane Proteins to Cilia. Cell (2010) 141(7):1208–19. doi:10.1016/j.cell.2010.05.015
11. OuBlacque, GO, E. Blacque, O, Snow, JJ, Leroux, MR, and Scholey, JM. Functional Coordination of Intraflagellar Transport Motors. Nature (2005) 436(7050):583–7. doi:10.1038/nature03818
12. Lechtreck, K-F, Johnson, EC, Sakai, T, Cochran, D, Ballif, BA, Rush, J, et al. The Chlamydomonas Reinhardtii BBSome Is an IFT Cargo Required for export of Specific Signaling Proteins from Flagella. J Cel Biol (2009) 187(7):1117–32. doi:10.1083/jcb.200909183
13. Ball, AD, Stapley, J, Dawson, DA, Birkhead, TR, Burke, T, and Slate, J. A Comparison of SNPs and Microsatellites as Linkage Mapping Markers: Lessons from the Zebra Finch (Taeniopygia guttata). BMC Genomics (2010) 11(1):218. doi:10.1186/1471-2164-11-218
14. Schönauer, R, Baatz, S, Nemitz-Kliemchen, M, Frank, V, Petzold, F, Sewerin, S, et al. Matching Clinical and Genetic Diagnoses in Autosomal Dominant Polycystic Kidney Disease Reveals Novel Phenocopies and Potential Candidate Genes. Genet Med (2020) 22(8):1374–83. doi:10.1038/s41436-020-0816-3
15. Lu, H, Galeano, MCR, Ott, E, Kaeslin, G, Kausalya, PJ, Kramer, C, et al. Mutations in DZIP1L, Which Encodes a Ciliary-Transition-Zone Protein, Cause Autosomal Recessive Polycystic Kidney Disease. Nat Genet (2017) 49(7):1025–34. doi:10.1038/ng.3871
16. Ott, J, Wang, J, and Leal, SM. Genetic Linkage Analysis in the Age of Whole-Genome Sequencing. Nat Rev Genet (2015) 16(5):275–84. doi:10.1038/nrg3908
17. Arélin, M, Schulze, B, Müller-Myhsok, B, Horn, D, Diers, A, Uhlenberg, B, et al. Genome-wide Linkage Analysis Is a Powerful Prenatal Diagnostic Tool in Families with Unknown Genetic Defects. Eur J Hum Genet (2013) 21(4):367–72.
18. Smith, KR, Bromhead, CJ, Hildebrand, MS, Shearer, AE, Lockhart, PJ, Najmabadi, H, et al. Reducing the Exome Search Space for Mendelian Diseases Using Genetic Linkage Analysis of Exome Genotypes. Genome Biol (2011) 12(9):R85–9. doi:10.1186/gb-2011-12-9-r85
19. Heyer, CM, Sundsbak, JL, Abebe, KZ, Chapman, AB, Torres, VE, Grantham, JJ, et al. Predicted Mutation Strength of Nontruncating PKD1 Mutations Aids Genotype-Phenotype Correlations in Autosomal Dominant Polycystic Kidney Disease. Jasn (2016) 27(9):2872–84. doi:10.1681/asn.2015050583
20. Bitarafan, F, and Garshasbi, M, Molecular Genetic Analysis of Polycystic Kidney Disease 1 and Polycystic Kidney Disease 2 Mutations in Pedigrees with Autosomal Dominant Polycystic Kidney Disease. J Res Med Sci (2019).24:44. doi:10.4103/jrms.JRMS_835_18
21. Likar, IP, Jere, KS, Možina, T, Verdenik, I, and Tul, N. Pregnancy Loss after Amniocentesis and Chorionic Villus Sampling: Cohort Study. Slovenian J Public Health (2020) 60(1):25–9. doi:10.2478/sjph-2021-0005
22. Cornec-Le Gall, E, Olson, RJ, Besse, W, Heyer, CM, Gainullin, VG, Smith, JM, et al. Monoallelic Mutations to DNAJB11 Cause Atypical Autosomal-Dominant Polycystic Kidney Disease. Am J Hum Genet (2018) 102(5):832–44. doi:10.1016/j.ajhg.2018.03.013
23. Shao, A, Chan, SC, and Igarashi, P. Role of Transcription Factor Hepatocyte Nuclear Factor-1β in Polycystic Kidney Disease. Cell Signal (2020) 71:109568. doi:10.1016/j.cellsig.2020.109568
24. Porath, B, Gainullin, VG, Cornec-Le Gall, E, Dillinger, EK, Heyer, CM, Hopp, K, et al. Mutations in GANAB , Encoding the Glucosidase IIα Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am J Hum Genet (2016) 98(6):1193–207. doi:10.1016/j.ajhg.2016.05.004
25. Richards, T, Modarage, K, Dean, C, McCarthy-Boxer, A, Hilton, H, Esapa, C, et al. Atmin Modulates Pkhd1 Expression and May Mediate Autosomal Recessive Polycystic Kidney Disease (ARPKD) through Altered Non-canonical Wnt/planar Cell Polarity (PCP) Signalling. Biochim Biophys Acta (Bba) - Mol Basis Dis (2019) 1865(2):378–90. doi:10.1016/j.bbadis.2018.11.003
26. Ali, H, Al-Mulla, F, Hussain, N, Naim, M, Asbeutah, AM, AlSahow, A, et al. PKD1 Duplicated Regions Limit Clinical Utility of Whole Exome Sequencing for Genetic Diagnosis of Autosomal Dominant Polycystic Kidney Disease. Sci Rep (2019) 9(1):4141–3. doi:10.1038/s41598-019-40761-w
27. Lu, Y, Peng, H, Jin, Z, Cheng, J, Wang, S, Ma, M, et al. Preimplantation Genetic Diagnosis for a Chinese Family with Autosomal Recessive Meckel-Gruber Syndrome Type 3 (MKS3). PloS one (2013) 8(9):e73245. doi:10.1371/journal.pone.0073245
28. Calzolari, E, Barisic, I, Loane, M, Morris, J, Wellesley, D, Dolk, H, et al. Epidemiology of Multiple Congenital Anomalies in Europe: A EUROCAT Population-Based Registry Study. Birth Defects Res A: Clin Mol Teratology (2014) 100(4):270–6. doi:10.1002/bdra.23240
29. Martinhago, C, Vagnini, L, Petersen, C, Mauri, A, Baruffi, R, De Oliveira, R, et al. Development of a Real-Time PCR Method for Rapid Sexing of Human Preimplantation Embryos. Reprod Biomed Online (2010) 20(1):75–82. doi:10.1016/j.rbmo.2009.10.008
30. Skrzypek, H, and Hui, L. Noninvasive Prenatal Testing for Fetal Aneuploidy and Single Gene Disorders. Best Pract Res Clin Obstet Gynaecol (2017) 42:26–38. doi:10.1016/j.bpobgyn.2017.02.007
31. Kutyavin, IV, Afonina, IA, Mills, A, Gorn, VV, Lukhtanov, EA, Belousov, ES, et al. 3'-Minor Groove Binder-DNA Probes Increase Sequence Specificity at PCR Extension Temperatures. Nucleic Acids Res (2000) 28(2):655–61. doi:10.1093/nar/28.2.655
32. Halbritter, J, Diaz, K, Chaki, M, Porath, JD, Tarrier, B, Fu, C, et al. High-throughput Mutation Analysis in Patients with a Nephronophthisis-Associated Ciliopathy Applying Multiplexed Barcoded Array-Based PCR Amplification and Next-Generation Sequencing. J Med Genet (2012) 49(12):756–67. doi:10.1136/jmedgenet-2012-100973
33. Mochizuki, T, Teraoka, A, Akagawa, H, Makabe, S, Akihisa, T, Sato, M, et al. Mutation Analyses by Next-Generation Sequencing and Multiplex Ligation-dependent Probe Amplification in Japanese Autosomal Dominant Polycystic Kidney Disease Patients. Clin Exp Nephrol (2019) 23(8):1022–30. doi:10.1007/s10157-019-01736-3
34. Simms, RJ, Eley, L, and Sayer, JA. Nephronophthisis. Eur J Hum Genet (2009) 17(4):406–16. doi:10.1038/ejhg.2008.238
35. Borràs, DM, Vossen, RHAM, Liem, M, Buermans, HPJ, Dauwerse, H, van Heusden, D, et al. Detecting PKD1 Variants in Polycystic Kidney Disease Patients by Single-Molecule Long-Read Sequencing. Hum Mutat (2017) 38(7):870–9. doi:10.1002/humu.23223
36. Moldován, N, Szűcs, A, Tombácz, D, Balázs, Z, Csabai, Z, Snyder, M, et al. Multiplatform Next-Generation Sequencing Identifies Novel RNA Molecules and Transcript Isoforms of the Endogenous Retrovirus Isolated from Cultured Cells. FEMS Microbiol Lett (2018) 365(5):fny013.
37. Samorodnitsky, E, Jewell, BM, Hagopian, R, Miya, J, Wing, MR, Lyon, E, et al. Evaluation of Hybridization Capture versus Amplicon‐Based Methods for Whole‐Exome Sequencing. Hum Mutat (2015) 36(9):903–14. doi:10.1002/humu.22825
38. Xing, D-J, Zhang, H-X, Huang, N, Wu, K-C, Huang, X-F, Huang, F, et al. Comprehensive Molecular Diagnosis of Bardet-Biedl Syndrome by High-Throughput Targeted Exome Sequencing. PLoS One (2014) 9(3):e90599. doi:10.1371/journal.pone.0090599
39. Ng, SB, Turner, EH, Robertson, PD, Flygare, SD, Bigham, AW, Lee, C, et al. Targeted Capture and Massively Parallel Sequencing of 12 Human Exomes. Nature (2009) 461(7261):272–6. doi:10.1038/nature08250
40. Dehghani, M, Mojarad, M, Ghayoor Karimiani, E, Vahidi Mehrjardi, MY, Sahebalzamani, A, Ashrafzadeh, F, et al. A Common Ancestral Asn242Ser Mutation in TMEM67 Identified in Multiple Iranian Families with Joubert Syndrome. Public health genomics (2017) 20(3):188–93. doi:10.1159/000477560
41. Mallawaarachchi, AC, Hort, Y, Cowley, MJ, McCabe, MJ, Minoche, A, Dinger, ME, et al. Whole-genome Sequencing Overcomes Pseudogene Homology to Diagnose Autosomal Dominant Polycystic Kidney Disease. Eur J Hum Genet (2016) 24(11):1584–90. doi:10.1038/ejhg.2016.48
42. Miller, NA, Farrow, EG, Gibson, M, Willig, LK, Twist, G, Yoo, B, et al. A 26-hour System of Highly Sensitive Whole Genome Sequencing for Emergency Management of Genetic Diseases. Genome Med (2015) 7(1):100–6. doi:10.1186/s13073-015-0221-8
43. Watson, CM, Dean, P, Camm, N, Bates, J, Carr, IM, Gardiner, CA, et al. Long‐read Nanopore Sequencing Resolves a TMEM231 Gene Conversion Event Causing Meckel-Gruber Syndrome. Hum Mutat (2020) 41(2):525–31. doi:10.1002/humu.23940
Keywords: whole exome sequencing, cilia, rare disease, polycystic kidney disease, ciliopathies
Citation: Modarage K, Malik SA and Goggolidou P (2022) Molecular Diagnostics of Ciliopathies and Insights Into Novel Developments in Diagnosing Rare Diseases. Br J Biomed Sci 79:10221. doi: 10.3389/bjbs.2021.10221
Received: 12 November 2021; Accepted: 02 December 2021;
Published: 10 January 2022.
Copyright © 2022 Modarage, Malik and Goggolidou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: P. Goggolidou, cC5nb2dnb2xpZG91QHdsdi5hYy51aw==