Abstract
Dystonia refers to a heterogeneous group of movement disorders characterized by involuntary, sustained muscle contractions leading to repetitive twisting movements and abnormal postures. Dystonia has a broad clinical spectrum and can affect different body regions, causing significant disability and reduced quality of life. Despite significant progress in understanding the disorder, many challenges in dystonia research remain. This mini-review aims to highlight the major challenges facing basic and translational research in this field, including 1) heterogeneity of the disorder, 2) limited understanding of its pathophysiology, 3) complications of using animal models, 4) lack of a framework linking genes, biochemistry, circuits, and clinical phenomenology, and 5) limited research funding. Identifying and discussing these challenges can help prioritize research efforts and resources, highlight the need for further investigation and funding, and inspire action towards addressing these challenges.
Introduction
The complexity of dystonia poses significant challenges to researchers striving to unravel its underlying mechanisms. One of the main challenges arises from the inherent heterogeneity of dystonia. With diverse forms of the disorder exhibiting variations in etiology, symptomology and associated features, a fundamental question emerges: should all types of dystonia be approached independently in terms of research, analysis and treatment, or they share an underlying biological pathway (1)? The limited number of well-documented pathological cases available to study the structural and anatomical features especially of isolated dystonia represents another significant challenge for researchers, who are eager to uncover the neurodevelopmental changes induced by dystonia-causing mutations in brain circuits involved in motor control and discern whether dystonia predominantly involves dysfunction in the brainstem, cerebellum, basal ganglia, or all [1–3]. In the past decade, in the attempt to elucidate dystonia pathogenesis and to facilitate the discovery of potential novel treatments, much effort has been dedicated to the development and characterization of animal models, and particularly rodent models of inherited dystonia. While instrumental for the molecular, cellular and circuit analyses in dystonia research, these animal models often fail to fully replicate the motor phenotype observed in humans [4]. This limitation presents a hurdle in studying the disorder under controlled conditions and testing novel therapeutic approaches. Connecting different biological levels of analysis poses yet another challenge in dystonia research [5]. Researchers strive to establish a comprehensive framework that links genetic mutations to alterations in brain structures, neurotransmitter systems, circuits, and ultimately the manifestation of clinical phenomena. Understanding how genetic mutations associated with dystonia impact the intricate interplay between neurotransmitter systems in movement-related brain regions is a vital avenue for investigation. Lastly, limited funding presents a significant obstacle in advancing dystonia research. Due to its relative rarity, securing sufficient funding for research studies becomes challenging.
This review aims to shed light on the complex landscape confronted by researchers in the field of pre-clinical dystonia research (Figure 1). The challenges they face are multifaceted and intricate, and addressing them will be instrumental in advancing our knowledge of dystonia, ultimately paving the way for improved diagnosis, treatment, and patient care.
FIGURE 1
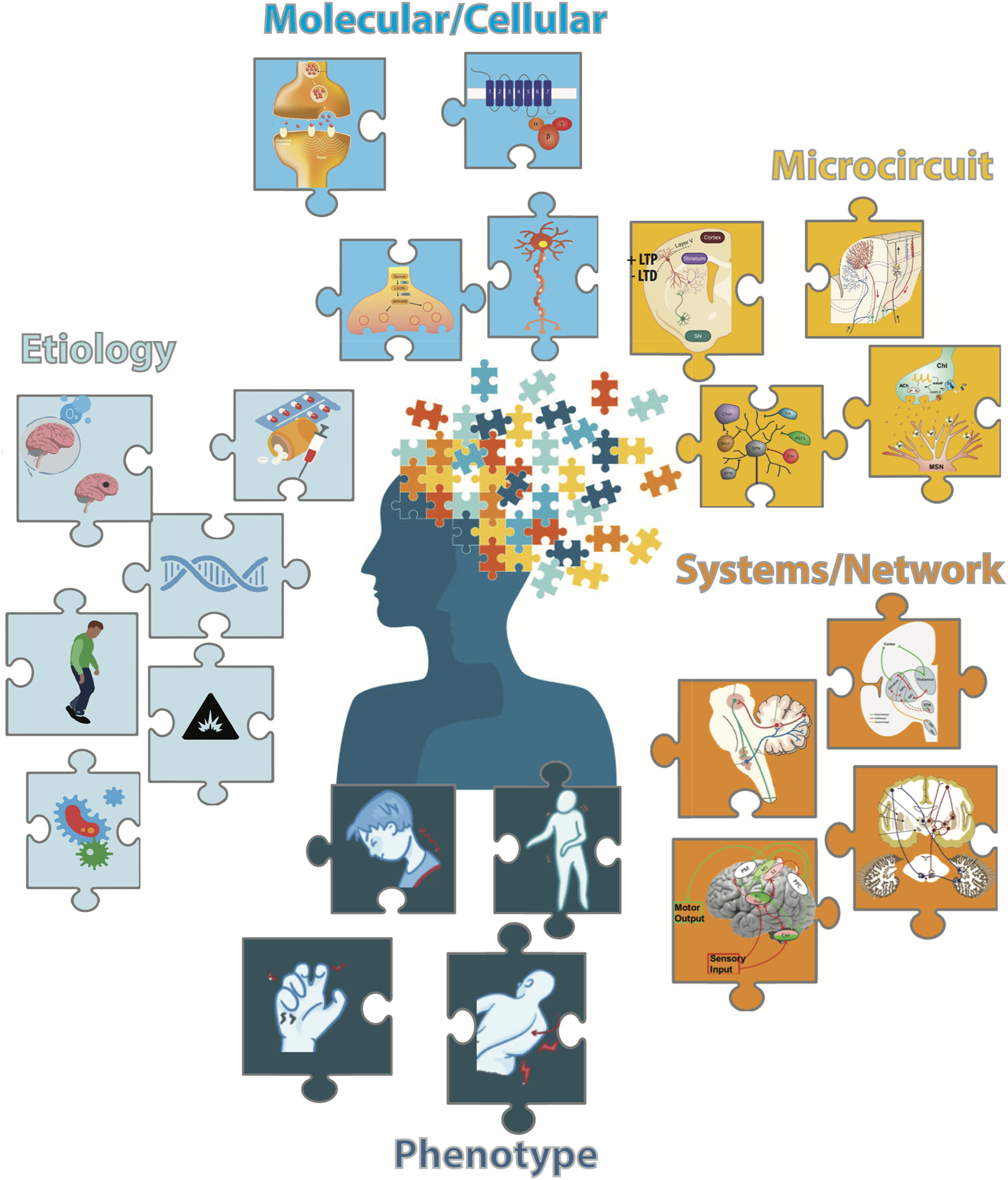
The multifaceted nature of dystonia is illustrated as different colored puzzle pieces: (Light Blue) Etiologies of Dystonia include brain tumors or hypoxia, prescription or recreational drugs, genetic factors, comorbidity with Parkinson's disease or other movement disorders, environmental factors and infections. (Cyan) Molecular/Cellular Dysfunctions underlying dystonia include synaptic functions/plasticity, abnormalities in signaling pathways, disruptions of neurotransmitter synthesis and neuronal functions.(Yellow) Microcircuit Dysfunctions include maladaptive synaptic plasticity at corticostriatal synapses, disruptions in the cerebellar microcircuit and imbalances in striatal neurotransmitter/neuromodulators systems. (Orange) System/Network Dysfunctions include the basal ganglia-thalamo-cortical and cerebello-thalamocortical circuits of dystonia, connections between the basal ganglia and cerebellum networks with potential interaction regions in dystonia including the thalamus and pontine nuclei (adapted from [6]) and brain network involved in sensory-motor integration with impaired connections between multisensory areas and M1 in the dystonic brain (adapted from [7]). (Dark Blue) Clinical Manifestations including cervical dystonia, generalized dystonia, hand dystonia (writer's cramp), and axial dystonia.
Heterogeneity of the disorder
Dystonia is a multifaceted disorder, with complex and heterogeneous etiologies, symptomologies, and courses [8]. The clinical presentation is highly variable and can range from isolated dystonia to dystonia as a comorbidity alongside other multi-systemic disorders. The complexity of the disease is compounded by its variability in virtually every measurable facet, including age of onset, body distribution, temporal pattern, and associated features in the clinical presentation.
Based on the body region affected, dystonia can be classified into 1) generalized dystonia, characterized by involuntary muscle contractions that affect the entire body, 2) segmental dystonia, affecting adjacent body regions, and 3) focal dystonia, limited to a specific body region, such as the neck (cervical dystonia), hand (writer’s cramp), or voice (spasmodic dysphonia). Each of these can present as isolated, combined, or complex forms of dystonia.
Etiology is also diverse, with various pathological findings, inheritance patterns (idiopathic, acquired, and inherited forms), and modes of acquisition. For instance, dystonia can be inherited in an autosomal dominant or recessive manner, or acquired as a result of trauma, infection, or exposure to medications or toxins [9]. This heterogeneity poses a challenge for research because it complicates identification of common mechanisms underlying dystonia and impedes development of effective treatments.
A fundamental, remaining question is whether different types of dystonia merely share a superficial phenotypic resemblance with entirely unrelated etiologies or whether shared biological substrates converge on an underlying biological pathway. Shared pathways may exist at the molecular, cellular, or anatomical level [10]. The discovery of common signaling mechanisms creates new opportunities for understanding the mechanisms of dystonia and developing treatment.
To tackle this question, research has focused on the study of specific genetic mutations associated with different types of dystonia. More than 300 genes capable of causing dystonia are known. These genetic mutations provide insights into the underlying molecular mechanisms [11]. At first glance, the functions of the dystonia genes appear quite divergent since they span a wide variety of biological processes including basic metabolic processes, cellular handling of ions, DNA transcription and repair, mitochondrial function, protein folding and trafficking, among others. Despite this divergent list, some common themes have been recognized and include defects in the structure of the basal ganglia and cerebellum, as well as altered dopamine signaling involved in movement control [10].
However, there is a significant variation in penetrance among dystonia-causing mutation carriers and in animal models recapitulating these mutations. This suggests that other factors, such as environmental and epigenetic factors and compensatory mechanisms, may play a role in the development of dystonia [12, 13]. Comorbidities, such as Parkinson’s disease, dyskinesia, or Huntington’s disease, can also contribute to the heterogeneity of dystonia, making it even more challenging to discern dystonia features and underlying mechanisms [14]. Understanding these factors is critical for the development of genetic animal models that more reliably express dystonic motor phenotypes and can be used to explore shared cellular pathophysiologic mechanisms. The ability to classify various forms of dystonia into subgroups based on cellular mechanisms is a crucial step in predicting which populations will benefit from new therapeutics targeting the disease mechanism. This research could also provide a convergent common target for symptomatic therapies across dystonia subtypes.
Limited understanding of pathophysiology
Dystonia is a multifactorial disorder resulting from a complex interplay between genetic, environmental, and neurochemical factors. Despite significant advances in identifying these factors, the identification of characteristic neuropathologic lesions associated with dystonia has been mostly elusive, challenging the understanding of the underlying mechanisms of dystonia. One hypothesis of the pathogenesis of dystonia is that multiple diverse intracellular defects resulting from different genetic and neuroplastic events may result in common abnormalities at the level of brain circuitry, thus producing a similar phenotype [3].
A core feature defining dystonia is co-contraction of agonist and antagonist muscle groups and overflow of activity into adjacent muscles. As such, dystonia can be considered a defect in motor control that involves matching the strength and distribution of muscles recruited to perform a specific task and coordinating the activity of agonists and antagonists for a particular movement. Understanding the circuit abnormalities underlying this defect requires fundamental knowledge regarding how the brain controls movement.
One approach to understanding the circuit abnormalities in dystonia is a striatal-centric view, grounded in ideas about the direct and indirect efferent pathways of striatal neurons. These models were developed largely from experimental data in Parkinson’s and Huntington’s disease models. Drawing from these disorders, it has been proposed that dystonia results from imbalanced activation of direct versus indirect striatal output pathways, ultimately releasing thalamo-cortical neurons from inhibition and promoting movement [15]. However, the exact mechanisms underlying this imbalance are still unclear. One hypothesis is that the critical element for striatal activity imbalance, and dystonia genesis, may be hyperactivity of striatal interneurons, particularly cholinergic [16, 17] and parvalbumin-positive interneurons [18, 19], which typically inhibit both dopamine D1 and D2 receptor-expressing striatal projection neurons (SPN) and have integral functions for regulation of motor focusing [20]. The excessive inhibition of SPNs could result in hyper-activation of the indirect pathway and hyper-deactivation of the direct pathway, leading to a loss of inhibition of “unwanted movement”. This ultimately could cause involuntary movements or agonist/antagonist muscle co-contractions [21, 22]. The amount of dopamine in the striatum could determine the scaling of abnormal movements: excessive dopamine amounts lead to enlarging movements via direct pathway facilitation, and reduced amounts of dopamine leading to reduced movements, that is fixed or tonic dystonia [22]. Although dysfunctions have been found in cholinergic and parvalbumin-positive interneurons in models of dystonia and dopamine signaling has been strongly implicated in several forms of dystonia, a cohesive and direct validation of this hypothesis is still lacking.
An alternative hypothesis hinges on evidence that the pathophysiology of dystonia may involve a combination of dysfunction within neurons of the brainstem, cerebellum, putamen, and globus pallidus [3]. Recently, more systematic neuropathological assessments associated with quantitative stereology-based measurements in human brain with isolated dystonia have found cellular abnormalities in the dopaminergic neurons of the substantia-nigra [23], in the cerebellar deep nuclei [24] and in the cholinergic neurons of the brainstem [25].
Growing evidence from basic and clinical research has elucidated the importance of the cerebellum, indicating that cerebellar abnormalities are present in individuals with dystonia [24, 26] and that inducing cerebellar dysfunction in animal models can produce dystonic symptoms [27–29]. This suggests that dystonia may arise from a motor network dysfunction, including the cortico-striato-pallido-thalamo-cortical and the cortico-ponto-cerebello-thalamo-cortical loops. These loops converge to regulate the activity of striatal cholinergic interneurons. As such, cholinergic interneuron dysfunction could represent a converging point of basal ganglia and cerebellar abnormality [22].
A less explored but intriguing hypothesis is that dystonia may result from aberrant motor engram formation. Motor engrams are the stored neural representations of motor movements [30]. Aberrant motor engrams may result in incorrect movement patterns [31], leading to dystonic symptoms. This hypothesis is supported by the assumption that dystonia is linked to the disruption of synaptic “scaling,” with a prevailing facilitation of synaptic potentiation, together with the loss of synaptic inhibitory processes at corticostriatal synapses described in multiple animal models and patients with dystonia [32].
In addition to genetic and neurochemical factors, inflammation and immune system dysfunction may also play a role in the development and progression of dystonia. Several studies have shown that inflammation and immune system dysfunction are present in individuals with cervical dystonia [33–35], suggesting that these factors may contribute to the pathophysiology of the disorder. However, the exact mechanisms underlying their role in dystonia remain unclear and understudied.
Limited understanding of the pathophysiology of dystonia poses a significant challenge in developing effective treatments for the disorder. In order to gain a better understanding of the underlying mechanisms of the disease, a prospective, quantitative study in well-phenotyped models with different types of genetic and isolated dystonia is necessary. Furthermore, a refinement of these pathophysiological notions is critically dependent on our capacity to discern and quantify specific movement abnormalities in different animal models and link them to neuronal activity in real-time. It is particularly important that this research is pursued in experimental models that allow for combining advanced behavioral analyses with molecular and systems-level investigations [36]. Emerging methodologies to track and characterize naturalistic behaviors at neural timescales and to monitor brain neurotransmission in vivo with unprecedented time resolution are now available and make the goal of linking circuit abnormalities to motor dysfunctions more achievable.
Limited animal models
Animal models are essential for understanding the underlying mechanisms of neurological disorders and developing potential treatments. Rodent models have been instrumental for elucidating the molecular, cellular and circuit aspects of dystonia, but limitations in their behavioral phenotype restrict the effectiveness in studying all aspects of the disorder. Dystonia animal models can be classified into two categories, etiological and symptomatic. Etiological mouse models of inherited dystonia reproduce the genetic mutations associated with the disorder. A large body of work utilizing either ubiquitous or conditional knock-out and knock-in approaches have revealed a myriad of biochemical, transcriptional, cellular and synaptic alterations in key brain areas controlling movement [37]. Some of these genetic models exhibit some level of abnormal movements at rest or with stimulation [38–43] but the majority do not display an overt dystonic phenotype [44]. Conversely, some symptomatic rodent models can at least partially mimic human dystonia phenotypes, though the etiology is either unknown or does not correspond to known etiologies in humans [44, 45]. These phenotypic models may be useful to test novel pharmacological agents and identify the anatomical and physiological processes involved. Establishment of a phenotypic model that is etiologically relevant to human dystonia and exhibits core behavioral characteristics including abnormal postures and agonist/antagonist co-contraction would be very valuable to the field.
Researchers have used a variety of approaches to define and characterize dystonic movements in animal models, most of which lack objectivity and/or temporal resolution. Electromyography (EMG) recordings have been used to show co-contraction of antagonist muscles [46], but these measurements are neither quantitative nor directly related to movement quality. Visual rating scales of dystonic and dyskinetic features have traditionally strongly relied on human-assigned labels and scoring, which lack not only objectivity but also temporal resolution. The development of unsupervised machine learning techniques for detection and monitoring of movements is important to enable progress in this research field. These technologies enable the capture of fine-grained behavioral patterns and identification of subtle differences between similar movements, making them the ideal candidates for detecting and characterizing abnormal postures and dystonic movements at neural timescales. These approaches could characterize dystonia in a non-biased, objective way in symptomatic mouse models (and humans) and could potentially identify previously unknown features of movement that may have been missed in the asymptomatic mouse models or carriers of dystonia-causing mutations. Moreover, characterization of complex motor behaviors at a sub-second timescale will help establish a correlation between dystonic movements and neural activity.
Lack of connection between different biological levels of analysis
One of the challenges of any research discipline is to integrate knowledge accumulated at several analytical levels. This fact is certainly relevant for dystonia research (Figure 1): It has been difficult to identify a framework for linking genes, biochemistry, circuits and clinical phenomena [5].
At the gene level, researchers have gained insights from studying both monogenic forms of dystonia, as well as other sporadic forms, by identifying specific genes associated with dystonia [9, 47]. This genetic information can uncover the molecular pathways and biological processes involved in the development and progression of the disorder but, for many dystonia genes, even this level of knowledge is still unclear.
At the biochemical level, abnormalities in neurotransmitter systems have been found to play a central role in dystonia. Dysfunctions in dopamine [48], acetylcholine [49], and GABAergic transmission [50], mainly in the basal ganglia, have been implicated in the pathophysiology of the disorder. However, how these dysfunctions arise from the genes involved in dystonia, how they impact neural circuits involved in the regulation of motor control and coordination and how this can lead to the manifestation of dystonic symptoms has not been fully elucidated.
At the circuit level, three common themes have been identified: loss of inhibition, abnormal sensorimotor integration, and maladaptive plasticity [5]. While these defects have been attributed to dysfunction of the basal ganglia, direct evidence for the responsible anatomical circuitry is lacking, and the possibility that they arise instead from dysfunction of the cerebellum [27, 28] or spinal neural circuits [41] has been raised. Evidence that these physiological defects play a causal role in dystonia also is lacking, and the possibility remains that some may be secondary to the movement disorder instead [51].
A network model of dystonia proposes that dysfunction occurs along the pathways connecting different brain regions, including the cortex, thalamus, basal ganglia, and cerebellum [52]. In dystonia, there is a disruption in the communication and coordination between these brain areas, resulting in abnormal motor output. Two independent papers found that dystonia-linked mutations in the gene THAP1 lead to impairment in myelin formation [53, 54], which could affect neuronal communication. The network model provides a framework for understanding how the dysfunction in these interconnected brain regions contributes to the development of dystonic symptoms.
Like other disorders, the pathogenesis of dystonia can be viewed as a multi-step process where some original insult triggers a series of abnormalities at the molecular, cellular, anatomical, and physiological levels [51]. Interactions among these levels may occur for specific subgroups of dystonia and pathogenesis of several subtypes could converge at one level [55]. By bridging the gap between genetic findings, biochemical abnormalities, circuit dysfunctions, and clinical manifestations, researchers can unravel the complex interplay between these factors and understand how different dystonias should be grouped. This integrated approach may necessitate a multidisciplinary team approach and will have important implications for experimental therapeutics.
Limited funding
Dystonia is a relatively rare disorder, affecting approximately 1% of the population. Compared to other neurological disorders, such as Alzheimer’s disease or Parkinson’s disease, dystonia receives limited funding. The only two available funding mechanisms specific to dystonia are currently the Dystonia Medical Research Foundation (DMRF) and Department of Defense (DoD) Peer-Reviewed Medical Research Program (PRMRP), which includes dystonia as one of the focus areas. The United States National Institutes of Health (NIH) reports spending $23M on dystonia research in 2022, but this includes a broad range of topics and is less than 1/10th the amount spent on Parkinson’s disease1.
This scarcity can limit the number of studies that can be conducted and the speed at which progress in understanding the disorder, developing new therapies, and conducting clinical trials can be made. The limited funding for dystonia research also affects the availability of resources, including animal models, equipment, and personnel. With limited funding, researchers may have to prioritize specific aspects of research, leading to gaps in knowledge and slow progress in the field.
Discussion
Dystonia is a complex disorder characterized by involuntary muscle contractions that result in abnormal, sustained movements and postures. Despite significant progress in understanding the disorder, many challenges in dystonia basic research still exist. The challenges discussed here, including heterogeneity, limited understanding of pathophysiology, limited animal models, lack of connection between different biological levels of analysis, and limited funding, are just some of the main difficulties encountered in this field. Addressing these challenges will require a multidisciplinary approach that involves collaboration between researchers, clinicians, and funding agencies. Innovative approaches, such as machine learning and modeling and sophisticated multi-site in vivo recordings of brain activity, may also be necessary to overcome these challenges and develop effective treatments for dystonia. This integrated approach is essential for identifying common pathways, shared mechanisms, and potential therapeutic targets that can lead to effective treatments for dystonia.
Statements
Author contributions
MS conducted the literature review, conceptualized and wrote the first draft of the manuscript, and designed the figure. DS reviewed and edited the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
Schirinzi T Sciamanna G Mercuri NB Pisani A . Dystonia as a network disorder: a concept in evolution. Curr Opin Neurol (2018) 31(4):498–503. 10.1097/WCO.0000000000000580
2.
Standaert DG . Update on the pathology of dystonia. Neurobiol Dis (2011) 42(2):148–51. 10.1016/j.nbd.2011.01.012
3.
Sharma N . Neuropathology of dystonia. Tremor Other Hyperkinet Mov (N Y) (2019) 9:569. 10.7916/d8-j6sx-b156
4.
Tassone A Sciamanna G Bonsi P Martella G Pisani A . Experimental models of dystonia. Int Rev Neurobiol (2011) 98:551–72. 10.1016/B978-0-12-381328-2.00020-1
5.
Lungu C Ozelius L Standaert D Hallett M Sieber BA Swanson-Fisher C et al Defining research priorities in dystonia. Neurology (2020) 94(12):526–37. 10.1212/WNL.0000000000009140
6.
Berman BD Jinnah HA . Dystonia: five new things. Neurol Clin Pract (2015) 5(3):232–40. 10.1212/CPJ.0000000000000128
7.
Avanzino L Tinazzi M Ionta S Fiorio M . Sensory-motor integration in focal dystonia. Neuropsychologia (2015) 79:288–300. 10.1016/j.neuropsychologia.2015.07.008
8.
Albanese A Sorbo FD Comella C Jinnah HA Mink JW Post B et al Dystonia rating scales: critique and recommendations. Mov Disord (2013) 28(7):874–83. 10.1002/mds.25579
9.
Grütz K Klein C . Dystonia updates: definition, nomenclature, clinical classification, and etiology. J Neural Transm (Vienna) (2021) 128(4):395–404. 10.1007/s00702-021-02314-2
10.
Jinnah HA Sun YV . Dystonia genes and their biological pathways. Neurobiol Dis (2019) 129:159–68. 10.1016/j.nbd.2019.05.014
11.
Keller Sarmiento IJ Mencacci NE . Genetic dystonias: update on classification and new genetic discoveries. Curr Neurol Neurosci Rep (2021) 21(3):8. 10.1007/s11910-021-01095-1
12.
Sciamanna G El Atiallah I Montanari M Pisani A . Plasticity, genetics and epigenetics in dystonia: an update. Handb Clin Neurol (2022) 184:199–206. 10.1016/B978-0-12-819410-2.00011-4
13.
Rauschenberger L Knorr S Pisani A Hallett M Volkmann J Ip CW . Second hit hypothesis in dystonia: dysfunctional cross talk between neuroplasticity and environment?Neurobiol Dis (2021) 159:105511. 10.1016/j.nbd.2021.105511
14.
Stephen CD Dy-Hollins M Gusmao CM Qahtani XA Sharma N . Dystonias: clinical recognition and the role of additional diagnostic testing. Semin Neurol (2023) 43(1):17–34. 10.1055/s-0043-1764292
15.
Wichmann T . Pathophysiologic basis of movement disorders. Prog Neurol Surg (2018) 33:13–24. 10.1159/000480718
16.
Bonsi P Cuomo D Martella G Madeo G Schirinzi T Puglisi F et al Centrality of striatal cholinergic transmission in Basal Ganglia function. Front Neuroanat (2011) 5:6. 10.3389/fnana.2011.00006
17.
Eskow Jaunarajs KL Scarduzio M Ehrlich ME McMahon LL Standaert DG . Diverse mechanisms lead to common dysfunction of striatal cholinergic interneurons in distinct genetic mouse models of dystonia. J Neurosci (2019) 39(36):7195–205. 10.1523/JNEUROSCI.0407-19.2019
18.
Bode C Richter F Spröte C Brigadski T Bauer A Fietz S et al Altered postnatal maturation of striatal GABAergic interneurons in a phenotypic animal model of dystonia. Exp Neurol (2017) 287(1):44–53. 10.1016/j.expneurol.2016.10.013
19.
Hamann M Richter A Meillasson FV Nitsch C Ebert U . Age-related changes in parvalbumin-positive interneurons in the striatum, but not in the sensorimotor cortex in dystonic brains of the dt(sz) mutant hamster. Brain Res (2007) 1150:190–9. 10.1016/j.brainres.2007.02.074
20.
Koós T Tepper JM . Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nat Neurosci (1999) 2(5):467–72. 10.1038/8138
21.
Mink JW . The basal ganglia: focused selection and inhibition of competing motor programs. Prog Neurobiol (1996) 50(4):381–425. 10.1016/s0301-0082(96)00042-1
22.
Morigaki R Miyamoto R Matsuda T Miyake K Yamamoto N Takagi Y . Dystonia and cerebellum: from bench to bedside. Life (Basel) (2021) 11(8):776. 10.3390/life11080776
23.
Iacono D Geraci-Erck M Peng H Rabin ML Kurlan R . Hypertrophy of nigral neurons in Torsin1A deletion (DYT1) carriers manifesting dystonia. Parkinsonism Relat Disord (2019) 58:63–9. 10.1016/j.parkreldis.2018.08.020
24.
Iacono D Peng H Rabin ML Kurlan R . Neuropathology and morphometry of dentate nucleus neurons in DYT1 brains: cerebellar abnormalities in isolated dystonia. J Neuropathol Exp Neurol (2023) 82(8):695–706. 10.1093/jnen/nlad044
25.
Mente K Edwards NA Urbano D Ray-Chaudhury A Iacono D Alho A et al Pedunculopontine nucleus cholinergic deficiency in cervical dystonia. Mov Disord (2018) 33(5):827–34. 10.1002/mds.27358
26.
Argyelan M Carbon M Niethammer M Ulug AM Voss HU Bressman SB et al Cerebellothalamocortical connectivity regulates penetrance in dystonia. J Neurosci (2009) 29(31):9740–7. 10.1523/JNEUROSCI.2300-09.2009
27.
Brown AM van der Heijden ME Jinnah HA Sillitoe RV . Cerebellar dysfunction as a source of dystonic phenotypes in mice. Cerebellum (2022) 22:719–29. 10.1007/s12311-022-01441-0
28.
Prudente CN Hess EJ Jinnah HA . Dystonia as a network disorder: what is the role of the cerebellum?Neuroscience (2014) 260:23–35. 10.1016/j.neuroscience.2013.11.062
29.
van der Heijden ME Kizek DJ Perez R Ruff EK Ehrlich ME Sillitoe RV . Abnormal cerebellar function and tremor in a mouse model for non-manifesting partially penetrant dystonia type 6. J Physiol (2021) 599(7):2037–54. 10.1113/JP280978
30.
Hwang FJ Roth RH Wu YW Sun Y Kwon DK Liu Y et al Motor learning selectively strengthens cortical and striatal synapses of motor engram neurons. Neuron (2022) 110(17):2790–801 e5. 10.1016/j.neuron.2022.06.006
31.
Girasole AE Lum MY Nathaniel D Bair-Marshall CJ Guenthner CJ Luo L et al A subpopulation of striatal neurons mediates levodopa-induced dyskinesia. Neuron (2018) 97(4):787–95. 10.1016/j.neuron.2018.01.017
32.
Quartarone A Pisani A . Abnormal plasticity in dystonia: disruption of synaptic homeostasis. Neurobiol Dis (2011) 42(2):162–70. 10.1016/j.nbd.2010.12.011
33.
Kilic-Berkmen G Scorr L Dinasarapu AR McKay L Rosen A Bagchi P et al Autoimmune and inflammatory mechanisms in cervical dystonia (2020). medRxiv.
34.
Panwar A Sawal N Garg D Kaur R . Idiopathic cervical dystonia – does autoimmunity play a hand?Mov Disord (2020) 35.
35.
Graziadio C Rosa RF Zen PR Pinto LL Barea LM Paskulin GA . Dystonia, autoimmune disease and cerebral white matter abnormalities in a patient with 18p deletion. Arq Neuropsiquiatr (2009) 67(3A):689–91. 10.1590/s0004-282x2009000400021
36.
Andreoli L Abbaszadeh M Cao X Cenci MA . Distinct patterns of dyskinetic and dystonic features following D1 or D2 receptor stimulation in a mouse model of parkinsonism. Neurobiol Dis (2021) 157:105429. 10.1016/j.nbd.2021.105429
37.
Rey Hipolito AG van der Heijden ME Sillitoe RV . Physiology of dystonia: animal studies. Int Rev Neurobiol (2023) 169:163–215. 10.1016/bs.irn.2023.05.004
38.
Rose SJ Yu XY Heinzer AK Harrast P Fan X Raike RS et al A new knock-in mouse model of l-DOPA-responsive dystonia. Brain (2015) 138(10):2987–3002. 10.1093/brain/awv212
39.
Nelson AB Girasole AE Lee HY Ptáček LJ Kreitzer AC . Striatal indirect pathway dysfunction underlies motor deficits in a mouse model of paroxysmal dyskinesia. J Neurosci (2022) 42(13):2835–48. 10.1523/JNEUROSCI.1614-20.2022
40.
Knorr S Rauschenberger L Pasos UR Friedrich MU Peach RL Grundmann-Hauser K et al The evolution of dystonia-like movements in TOR1A rats after transient nerve injury is accompanied by dopaminergic dysregulation and abnormal oscillatory activity of a central motor network. Neurobiol Dis (2021) 154:105337. 10.1016/j.nbd.2021.105337
41.
Pocratsky AM Nascimento F Özyurt MG White IJ Sullivan R O'Callaghan BJ et al Pathophysiology of Dyt1-Tor1a dystonia in mice is mediated by spinal neural circuit dysfunction. Sci Transl Med (2023) 15(694):eadg3904. 10.1126/scitranslmed.adg3904
42.
Pappas SS Li J LeWitt TM Kim JK Monani UR Dauer WT . A cell autonomous torsinA requirement for cholinergic neuron survival and motor control. Elife (2018) 7:e36691. 10.7554/eLife.36691
43.
Aïssa HB Sala RW Georgescu Margarint EL Frontera JL Varani AP Menardy F et al Functional abnormalities in the cerebello-thalamic pathways in a mouse model of DYT25 dystonia. Elife (2022) 11:e79135. 10.7554/eLife.79135
44.
Meringolo M Tassone A Imbriani P Ponterio G Pisani A . Dystonia: are animal models relevant in therapeutics?Rev Neurol (Paris) (2018) 174(9):608–14. 10.1016/j.neurol.2018.07.003
45.
Richter F Richter A . Genetic animal models of dystonia: common features and diversities. Prog Neurobiol (2014) 121:91–113. 10.1016/j.pneurobio.2014.07.002
46.
Scholle HC Jinnah HA Arnold D Biedermann FH Faenger B Grassme R et al Kinematic and electromyographic tools for characterizing movement disorders in mice. Mov Disord (2010) 25(3):265–74. 10.1002/mds.22933
47.
Zech M Jech R Boesch S Škorvánek M Weber S Wagner M et al Monogenic variants in dystonia: an exome-wide sequencing study. Lancet Neurol (2020) 19(11):908–18. 10.1016/S1474-4422(20)30312-4
48.
Ribot B Aupy J Vidailhet M Mazère J Pisani A Bezard E et al Dystonia and dopamine: from phenomenology to pathophysiology. Prog Neurobiol (2019) 182:101678. 10.1016/j.pneurobio.2019.101678
49.
Eskow Jaunarajs KL Bonsi P Chesselet MF Standaert DG Pisani A . Striatal cholinergic dysfunction as a unifying theme in the pathophysiology of dystonia. Prog Neurobiol (2015) 127-128:91–107. 10.1016/j.pneurobio.2015.02.002
50.
Lizarraga KJ Gorgulho A Chen W De Salles AA . Molecular imaging of movement disorders. World J Radiol (2016) 8(3):226–39. 10.4329/wjr.v8.i3.226
51.
Jinnah HA Berardelli A Comella C Defazio G Delong MR Factor S et al The focal dystonias: current views and challenges for future research. Mov Disord (2013) 28(7):926–43. 10.1002/mds.25567
52.
Jinnah HA Neychev V Hess EJ . The anatomical basis for dystonia: the motor network model. Tremor Other Hyperkinet Mov (N Y) (2017) 7:506. 10.7916/D8V69X3S
53.
Yellajoshyula D Rogers AE Kim AJ Kim S Pappas SS Dauer WT . A pathogenic DYT-THAP1 dystonia mutation causes hypomyelination and loss of YY1 binding. Hum Mol Genet (2022) 31(7):1096–104. 10.1093/hmg/ddab310
54.
Domingo A Yadav R Shah S Hendriks WT Erdin S Gao D et al Dystonia-specific mutations in THAP1 alter transcription of genes associated with neurodevelopment and myelin. Am J Hum Genet (2021) 108(11):2145–58. 10.1016/j.ajhg.2021.09.017
55.
Gonzalez-Latapi P Marotta N Mencacci NE . Emerging and converging molecular mechanisms in dystonia. J Neural Transm (Vienna) (2021) 128(4):483–98. 10.1007/s00702-020-02290-z
Summary
Keywords
dystonia, animal models, basic research, pathophysiology, translational research
Citation
Scarduzio M and Standaert DG (2023) Piecing together a complex puzzle: 5 key challenges in basic dystonia research. Dystonia 2:11615. doi: 10.3389/dyst.2023.11615
Received
25 May 2023
Accepted
11 December 2023
Published
21 December 2023
Volume
2 - 2023
Edited by
Aasef Shaikh, Case Western Reserve University, United States
Updates
Copyright
© 2023 Scarduzio and Standaert.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariangela Scarduzio, scardu@uabmc.edu
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.