 Lauren N. Miterko-Myers
Lauren N. Miterko-Myers- Peter O’Donnell Jr. Brain Institute, University of Texas Southwestern Medical Center, Dallas, TX, United States
Dystonia is a neurodevelopmental disorder characterized by severe involuntary twisting movements, hypothesized to arise from a dysfunctional motor network involving the cortex, basal ganglia, and cerebellum. Within this network, striatal cholinergic interneurons have been identified as possible contributors to dystonia pathophysiology. However, little is known about striatal cholinergic interneuron development in the mammalian brain, limiting our understanding of its role in dystonia and therapeutic potential. Here, I review striatal cholinergic interneuron development in the context of early-onset DYT1 (or “DYT-TOR1A”) dystonia. I discuss clinical and laboratory research findings that support cholinergic dysfunction in DYT1 dystonia and the implications of abnormal cholinergic cell development on disease penetrance and striatal connectivity.
Introduction
Dystonia is the third most common movement disorder in the United States, manifesting as abnormal, disabling postures in children, adolescents, and adults [1]. Dystonia is classified as either isolated (“primary”) or acquired (“secondary”) and can be inherited (e.g., DYT1) or caused by injury (e.g., stroke). A dominantly inherited mutation (
One way to overcome this challenge is by identifying cell types directly involved in DYT1 dystonia pathophysiology. The oral medications prescribed to patients following initial diagnoses—anticholinergics, benzodiazepines, levodopa, and antiepileptics—broadly alter cholinergic, GABAergic, dopaminergic, and glutamatergic cell activity and hint at possible contributors [8]. Trihexyphenidyl, an anticholinergic drug, is among the most effective treatments for DYT1 dystonia, especially in pediatric patients who exhibit greater tolerance to higher doses [8, 9]. Considered in conjunction with human neuroimaging studies that show an age-dependent disruption of striatal vesicular acetylcholine transporter (VAChT) expression in DYT1 patients [10], cholinergic neurons appear important early in dystonia pathophysiology. Whether these two phenotypes are physiologically connected is unclear but altered VAChT expression and cholinergic hyperactivity are not mutually exclusive given the functional redundancy of vesicular proteins in striatal cholinergic cells and co-expression of neurotransmitters [11–13]. Comprising 1%–3% of the cell population, cholinergic interneurons use acetylcholine (or glutamate) to modulate striatal circuit functions and motor control. Acetylcholine is loaded into pre-synaptic vesicles by VAChT and VGLUT3 and its release counterbalances the effects of dopamine on neuronal excitability and plasticity [11, 14]. During striatal development, acetylcholine modulates dopamine release to direct medium spiny neuron maturation and glutamatergic receptor expression [15, 16]. Early dopamine loss stunts medium spiny neuron growth, increases excitability, and impairs behavior [15, 17]. While the direct impact of cholinergic dysfunction on behavior has not been investigated in the juvenile striatum, it is likely that motor functions are compromised because cholinergic maturation overlaps with the development of locomotor-related movements and their loss or misfiring contributes to the motor deficits found in Parkinson’s disease, Huntington’s disease, and Tourette Syndrome patients [18–20].
A more thorough understanding of striatal cholinergic interneuron development may inform DYT1 origins and targeted therapies. In this mini review, I focus on the neurodevelopmental disorder of DYT1 dystonia, where I examine the evidence supporting cholinergic dysfunction in its pathophysiology. I then discuss what is known about striatal cholinergic interneuron development and compare its timeline to that of striatal maturation, with the intention of highlighting periods of motor circuit vulnerability applicable to dystonia. I will end considering maladaptation and implications of abnormal cholinergic cell development on DYT1 penetrance and treatments.
DYT1 dystonia as a neurodevelopmental disorder
Human patients and clinical observations have historically been used to characterize DYT1 dystonia etiology [21, 22]. Now, preclinical models support more in-depth investigations into DYT1 pathophysiology, including the identification of developmental alterations. Genetically engineered DYT1 mice generated through overexpressing human
Cholinergic dysfunction and DYT1 development
The plasticity alterations—enhanced long-term potentiation (LTP) and absent long-term depression (LTD)—previously discovered in postnatal DYT1 mice are restricted to the striatum, occurring specifically at corticostriatal synapses [28, 38]. Accompanying impaired plasticity is reduced dopamine binding to D1 and D2 receptors, which is critical for LTP and LTD induction [39, 40]. Not all DYT1 model mice exhibit impaired D1 and D2 receptor activity [41], lending support to the idea that input from other cells, such as striatal cholinergic interneurons, may be responsible for altering plasticity. The striatal cholinergic interneuron pathology that has been reported in DYT1 models with altered plasticity are cell degeneration, increased firing, and enhanced acetylcholine tone [29, 38, 42–47]. To determine whether hypercholinergic phenotypes contribute to DYT1 pathophysiology, antimuscarinics were administered before measuring plasticity and behavior [24, 38, 48]. Drugs, such as trihexyphenidyl, subdue cholinergic activity, improved motor symptoms, and normalized medium spiny neuron LTP and LTD in different DYT1 mouse models [24, 49]. These data call attention to the important role cholinergic neurons may play in DYT1 pathogenesis and unveil possible pharmacological mechanisms (i.e., plasticity restoration) for antimuscarinics in dystonia patients [50, 51].
From the above pharmacological and electrophysiological studies, it remains unclear whether the functional alterations found in striatal cholinergic cells are a cause of dystonia or consequence of torsinA mutations. Chemogenetic and transgenic mouse studies more directly test cholinergic interneuron involvement. Using Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) under the choline acetyltransferase (ChAT) promoter, Gemperli et al., (2022) found that chronic striatal cholinergic excitation incited dystonic behavior [52]. Abnormal motor behavior and responses to dopamine (D2/D3) receptor activation in the striatum also developed after deleting exons 3-4 of torsinA only in cholinergic cells using mice expressing Cre recombinase under the ChAT promoter [53]. Together, these cell-specific manipulations hint at a causal role of cholinergic interneurons in DYT1 dystonia, as do early intervention studies. Given that trihexyphenidyl is effective in a subset of children and that altered synaptic plasticity is found in some mouse models within 3 weeks of life, striatal cholinergic interneurons may contribute to DYT1 behavioral onset [8, 23]. Evidence suggesting early cholinergic involvement include: 1) striatal cholinergic interneurons degenerate just prior to motor symptoms manifesting in juvenile Dlx-CKO mice [24], 2) developing striatal Dlx-CKO cholinergic interneurons exhibit altered synaptic gene expression [37], and 3) striatal cholinergic interneurons demonstrate abnormal excitatory dopamine receptor responses in juvenile hMT mice [29]. Interestingly, these findings occur at P14-15, ages corresponding to neural and behavioral maturation in mice and approximately early adolescence and the average age-of-DYT1-onset in humans [8, 54, 55]. While fundamental developmental events in the striatum—including, rostral-to-caudal organization, cell identity, and striosome and matrix compartmentalization—have been extensively characterized from embryogenesis through P14 [56–60], the developmental trajectories of individual cholinergic neurons are less clear. One reason being the presumption of a structurally normal nervous system in DYT1 dystonia had stalled efforts to track anatomic changes [61–65]. Now, with advances in neuroimaging, microstructural changes throughout the adult nervous system have been identified [66, 67]. Detailed morphometric analyses in mature DYT1 tissue reveal enlarged striatal cholinergic cell bodies, postulated to result from altered connectivity [24, 68]. Repeating these experiments in developing DYT1 tissue will contribute to making a timeline of changes in dystonia.
Striatal cholinergic interneuron development and connectivity
Nearly all striatal cholinergic neurons are Gbx2-expressing cells born in the medial ganglionic eminence and preoptic area, that then undergo tangential migration [69]. A small proportion of striatal cholinergic neurons co-expressing Nkx2.1 and Zic4 are born in and have migrated from the septum [70]. Cholinergic interneurons are among the first striatal cell types to be born, between embryonic days (E) 12 and E17 in rat (E11.5-E14.5 in mouse; [57, 58]). In contrast, striatal medium spiny neurons are born as late as P0-P5 in rat [71–74]. By the end of the first postnatal week (P0-P6), striatal cholinergic neurons are postmitotic, express acetylcholinesterase (AChE), release acetylcholine, and start maturing morphologically [69–71, 75–78], raising the possibility of these cells playing a crucial role in circuit assembly. Providing further support for this hypothesis are the concurrent timelines for striatal cholinergic synaptogenesis, morphogenesis, and locomotor maturation [54, 79]. Excitatory afferents enter the striatum starting at E12, then significantly increase in density through the second postnatal week (Figures 1A, B; [80, 82]). Intrastriatal GABAergic connections form later, starting at P0-P4, with a density that remains constant throughout postnatal development (Figures 1A, B; [80]). The fluctuation in density of striatal excitatory inputs coincides cholinergic morphogenesis and developing locomotor behavior (Figures 1C, D). In rodents, excitatory innervation onto striatal cholinergic interneurons increases as locomotion increases and the dendritic arbor grows in size and complexity (Figures 1C, D; P7-13 [81, 83]) More specifically, movement begins at P8, when all striatal cell types are present and cell densities are comparable to that of adults [72]. Although circuit components are present and operational by P8, neuronal maturation is incomplete and locomotion is unrefined (Figures 1B, C; [54, 72, 74]). At P15, this changes when medium spiny and cholinergic neuronal counts and morphology become adult-like [72, 75, 81, 84]. Simultaneous excitatory synapse pruning and dendritic remodeling in the third postnatal week (P14-P20) overlaps with the acquisition of activity-dependent synaptic plasticity properties and the transformation of gait into that of adult rodents (Figures 1C, D; [79, 85, 86]). During the fourth postnatal week (P21-P27), gait maturation is complete, while striatal cholinergic morphology and connectivity undergoes fine-tuning (Figures 1C, D; [54, 85, 86]).
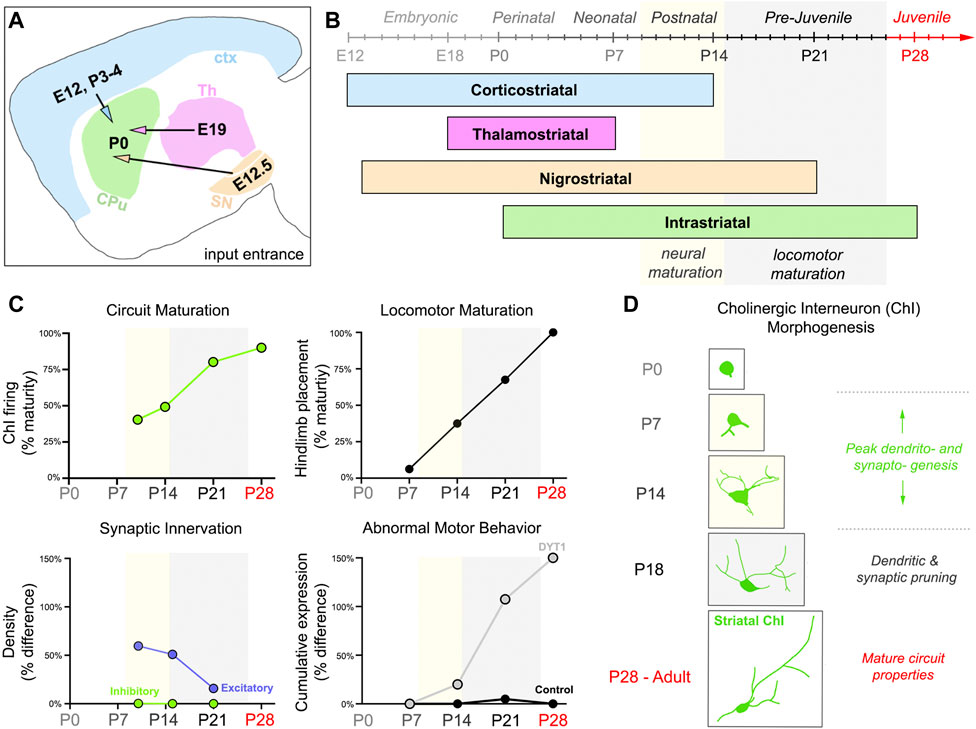
Figure 1. Striatal connectivity during development and locomotor maturation. (A) Schematic detailing the times (Embryonic day, E; Postnatal day, P) in which major excitatory afferents (ctx = cortex, blue; Th = thalamus, magenta; SN = substantia nigra, orange) project into the developing mouse striatum or are made internally (CPu = Caudate putamen, green). (B) Developmental timeline of striatal connectivity. Embryonic (E0–E20), perinatal (P0–P3), neonatal (P3–P9), postnatal (P9–P15), pre-juvenile (P15–P26), and juvenile (P26–P60) refer to periods of mouse behavioral development, as described in Fox, 1965 [54]. Mature neural properties emerge during the postnatal period (P9–P15, yellow). Locomotion becomes refined and more adult-like in pre-juvenile mice (P15–P26, gray). (C) Abnormal motor behaviors manifest postnatally in a symptomatic DYT1 mouse model, while striatal cholinergic interneuron (ChI) function, striatal innervation, and locomotion are maturing. Graphs were adapted by plotting calculations performed on data published in different sources. Circuit Maturation was calculated from data published by McGuirt et al., (2021) [81], licensed under CC BY 4.0. Locomotor Maturation was calculated from data published by Shriner et al., (2009) [86], with permission from Elsevier. Circuit and locomotor maturation estimates (0%–100%) were calculated using the formula: % maturity = (developmental measurement/adult measurement)*100%. Data included ChI firing (e.g., spontaneous firing frequency and coefficient of variation) and hindlimb placement measurements (i.e., the last locomotor feature to develop). A percent estimate of 100% represents complete maturation of the property studied. Synaptic innervation was measured by excitatory and inhibitory density values in the whole striatum, estimated from data published by Tepper et al., (1998) [80], with permission from Karger Publishers Percentages (0%–150%) represent the difference in innervation (excitatory, purple; inhibitory, green) between pups and adults. Limb clasping and grid hang failures were the abnormal motor behaviors summarized. Percentages (0%–150%) represent the cumulative expression of behaviors (Grid Hang % difference + Limb Clasping % difference) between control and Dlx-CKO mice. [24] Calculations were made using the limb clasping and grid hang failure data published in Pappas et al., (2015) [24], licensed under CC BY 4.0. Postnatal day P. (D) Striatal ChI morphology and connectivity co-evolve, with peak dendritic complexity and innervation exhibited by Postnatal day (P)14. Periods of neural (yellow) and locomotor (gray) maturation correspond to times of active ChI dendritic and synaptic remodeling in (C,D).
The final striatal circuit is achieved in mice approximately 1 month after birth (P28+), when medium spiny neuron spine density and excitability normalizes and cholinergic interneurons lose their globose shape, larger somata, and complex branching in favor of irregular shapes, fewer bifurcations, and dendrites spanning up to 1 mm (Figure 1D; [75, 79, 81, 87]). Cortical, thalamic, and dopaminergic afferents are also specifically organized on mature cholinergic interneurons. Corticostriatal synapses are the sparsest, and predominantly form on the distal dendrites of cholinergic interneurons [87–90]. In contrast, thalamostriatal synapses are the most abundant, concentrated on both the proximal dendrites and somata [87, 88, 90]. Dopaminergic afferents activate D1/D5 and D2 receptors, which are similarly localized throughout the cell, dispersed across the cholinergic somata and neurites [91–94]. The spatial distribution and quantity of each excitatory synapse type is not known for striatal cholinergic interneurons during postnatal development, but likely differs from adults, when dendritic remodeling and synaptic pruning is complete. Characterizing synaptic inputs onto striatal cholinergic interneurons throughout postnatal development may be important for understanding the pathogenesis of neurodevelopmental disorders, including DYT1 dystonia, especially given that torsinA is intensely expressed in striatal cholinergic interneurons from P14 to P21 and its functions have been implicated in the secretory pathway and synaptogenesis [37, 95, 96].
Altogether, several developmental processes must be coordinated within the mouse striatum during the first 4 weeks of life to establish a functional, mature circuit and execute movement. Whether striatal cholinergic synaptogenesis and morphogenesis directs behavioral changes remains to be elucidated, but their embryonic or conditional loss from the forebrain supports this prospect [97, 98]. Additionally, some DYT1 models (e.g., [24, 26, 27]) develop motor symptoms (P14-P21) as cholinergic neuron firing and morphology matures (Figures 1C,D; [79, 81]). During normal striatal development, cholinergic interneurons fire with increased frequency, pacemaking, and less irregularity from P14 to P18, as excitatory synaptic input and dendritic branching becomes refined [81, 99]. Prolonged synaptogenesis or impaired pruning resulting in aberrant excitatory connections onto striatal cholinergic interneurons may explain why some DYT1 models have enhanced acetylcholine tone or increased striatal cholinergic neuron excitability (e.g., [24, 29, 42, 44, 46]) and are responsive to anticholinergic interventions [24, 49]. Not only are cholinergic interneurons particularly sensitive to changes in excitatory input [88, 100, 101], but they also start making connections themselves, onto neighboring cells from P15 to P21, as evidenced by a drastic increase in background ChAT staining [75]. Therefore, increased cholinergic excitation would have downstream effects on medium spiny neuron function, with one effect being increased synchronization [102]. Human dystonia patients exhibit neuronal synchronization in the globus pallidus internus (GPi) due to burst-firing propagating from the striatum [102–106]. Burst-firing and neuronal synchronization is presumed to underlie dystonic postures because behavior improves when either are alleviated through GPi-targeted deep brain stimulation (GPi-DBS) or anticholinergics [106–109].
Discussion
Here, I considered the development of striatal cholinergic interneurons and their possible role in DYT1 pathogenesis. Not only is striatal cholinergic interneuron dysfunction detected in several DYT1 models, but cholinergic receptor expression is altered in patients and anticholinergic medications are among the leading treatment options to manage their motor symptoms [8, 10, 23, 24, 43, 44, 110]. However, not knowing the role of cholinergic interneurons in dystonia limits our understanding of antimuscarinic-based therapies and their efficacy in DYT1 patients. Given that DYT1 has neurodevelopmental origins and cholinergic interneurons are among the first striatal cell type to be born and reach functional maturity, it is possible that cholinergic interneurons are fundamentally involved in setting up a dysfunctional striatal circuit. This phenomenon is found in other motor regions and neurodevelopmental disorder models. For instance, Purkinje cells are among the first cell type to be born in the cerebellum (∼E10.5–E12.5), and instructs the development of granule cells, which constitute approximately 99% of the cerebellum via neurochemical cues [111–113]. Altering Purkinje cell functioning through pharmacological or genetic manipulations disrupts interactions with granule cells and leads to aberrant cerebellar circuit development and motor abnormalities [114–118]. In the postnatal striatum, cholinergic interneurons could direct innervation by excitatory afferents, which in turn prompts medium spiny neuron maturation and supports motor learning and execution. Cholinergic interneurons are in a position to coordinate rudimentary connections with excitatory projections because 1) cortical and dopaminergic afferents enter the striatum when cholinergic neurons are postmitotic and active, but medium spiny neurons are still being born [71, 74, 77]; 2) Cholinergic neurons act as “gatekeepers” and control excitatory inputs onto medium spiny neurons [119, 120]; 3) Cholinergic survival is unaffected by early dopaminergic or cortical denervation [121]. Indeed, cholinergic interneurons modulate the development of nigrostriatal pathways through expressing sonic hedgehog and TrkA, a nerve growth factor receptor [122, 123]. Both sonic hedgehog and TrkA signaling in the postnatal striatum maintains cholinergic cell numbers, which is required for nigrostriatal connectivity [122]. Through proper nigrostriatal connectivity, dopamine is released and guides medium spiny neuron maturation [15, 16, 81]. TrkA expression is downregulated in adult DYT1 patient and Dlx-CKO mouse model tissue [24], suggesting cholinergic dysfunction in dystonia. If TrkA signaling is dysregulated earlier, striatal connectivity could be impacted.
While altered striatal cholinergic interneuron instruction is possible, it is more likely that the striatal circuit develops normally but later becomes abnormal. Evidence for this hypothesis includes neonatal DYT1 mice exhibiting normal reflex behaviors [24]. When reflex behaviors develop normally, it suggests that underlying neural substrates are present and properly integrated into the motor circuit [54]. Later dysfunction of these neural substrates due to genetic insults, environmental stresses, or a combination of both then contributes to the development of abnormal behaviors [8, 124, 125]. In the Dlx-CKO mouse model where neonatal, but not postnatal, behaviors are preserved [24], the introduction of sensory inputs into the motor circuit may contribute to this shift. In the striatum, sensory inputs start influencing circuit function from P15 on, corresponding to a time of cortico-striatal plasticity changes [126] and locomotor behavior refinement [54]. Both medium spiny neurons and striatal cholinergic interneurons play integral roles in sensorimotor processing through their integration of dopaminergic and glutamatergic inputs [127]. This process is impaired in the hMT model, where plasticity or D2-receptor signaling defects were found in developing medium spiny neurons and striatal cholinergic interneurons [28, 29]. While similar observations have been reported in adult Tor1aΔGAG/+, Tor1a+/−, and muscarinic receptor (M2/M4) double knockout mice [38, 128, 129], determining how reproducible these electrophysiological phenotypes are in development requires earlier timepoints. Increased investigation into distinguishing the contributions of medium spiny neurons from striatal cholinergic interneurons in sensorimotor processing during development and across models are also necessary.
Interestingly, what makes developing striatal cholinergic interneurons individually equipped to integrate sensory inputs (e.g., their D2 and corticotropin releasing factor type 1 (CRF-R1) receptors), could also provide an explanation for the incomplete penetrance of DYT1 dystonia [79, 101]. Activation of D2 and CRF-R1 receptors enable reward as well as alters striatal cholinergic interneuron activity specifically in response to environmental cues [130, 131]. One prevailing hypothesis for the incomplete penetrance of DYT1 is that loss of torsinA primes the circuit for dysfunction, but symptoms only manifest if a secondary, external insult is applied [125]. Since striatal cholinergic interneurons normally express torsinA and can directly change their firing in response to environmental cues, then this two-hit hypothesis is plausible, despite not knowing the triggering sensory event(s). Another possibility is that DYT1 dystonia manifests when there is maladaptation or a lack of compensation. Focal dystonia is a prime example of maladaptation, where abnormal connectivity between the basal ganglia, cerebellum, and cortex develops as a result of repeatedly engaging muscles in prolonged motor tasks, such as writing or playing an instrument [132]. Unlike focal dystonia, maladaptation in DYT1 could result from changes occurring during development because peak torsinA expression coincides with periods of synaptogenesis in maturing striatal cells. Indeed, both striatal cholinergic interneurons and medium spiny neurons show evidence of increased synaptic inputs through exhibiting larger somata, increased membrane capacitances, and more spines [24, 37]. Alternatively, DYT1 dystonia could manifest due to a lack of compensation, which is supported by animal model studies. TorsinB is structurally and functionally similar to torsinA and can rescue cellular and behavioral phenotypes in Dlx-CKO mice, when overexpressed [133–136]. Reducing torsinB levels in Dlx-CKO animal models worsens DYT1 phenotypes, indicating that loss of torsinA drives dysfunction while torsinB attempts to normalize dysfunction [133]. Despite an early loss of torsinA (from ∼E11), the timing of torsinB expression remains the same (
Questions remain regarding how involved striatal cholinergic interneurons are in DYT1 pathophysiology, including: Are cholinergic neurons main contributors to dystonic postures? Are striatal cholinergic interneurons aberrantly connected? Are observations detailing striatal cholinergic dysfunction in adults conserved during development? Major limitations to answering these questions is a lack of behavioral reproducibility and consistency among cellular phenotypes across preclinical DYT1 models. Standardizing experimental readouts and testing various ages can help, but the lack of reproducibility also stems from the biological and technical requirements of different genetic models and an unclear understanding of torsinA mutations in cholinergic neurons. For example, some studies suggest that striatal cholinergic excitation alone is not sufficient to elicit dystonia-like behaviors [137, 138], but the duration of striatal cholinergic excitation may be imperative for behavioral manifestation [52]. As for our understanding of pathogenic torsinA mutations, they are hypothesized to result in a loss-of-function or dominant-negative effect. Not knowing which mechanism(s) torsinA mutations utilize—or which under what circumstances—affects how we develop models and interpret data. Accumulating evidence in human cells and DYT1 knockin (Tor1aΔGAG/ΔGAG, Tor1aΔGAG/+) mice suggests that the
Overall better functional characterizations will improve our understanding of dystonia, the cells contributing to its pathophysiology, and the clinical relevance of basic DYT1 research. This includes providing a timeline of the physiological changes surrounding dystonia onset because currently, there is no consensus on how early pathogenesis begins. Already, there have been advances in elucidating torsinA roles in nuclear-cytoplasmic communication [32, 147] and uniting behavior from preclinical and clinical work, including the finding that both human patients and dystonia animal models exhibit variable leg adduction [52]. Increased knowledge will inevitably guide the development of more effective therapies by improving target specificity and timing.
Author contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
I would like to thank the National Institute of Neurological Disorders and Stroke (NINDS; Bethesda, United States) for their funding of my postdoctoral work (F32NS128000).
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I would like to thank Kareena Arora for her help finding, organizing, and summarizing primary data articles for this mini review. I would also like to thank Dr. Samuel S. Pappas and Dr. Meike van der Heijden for their critical feedback.
References
1. Stephen, CD. The dystonias. Contin (Minneap Minn) (2022) 28(5):1435–75. doi:10.1212/CON.0000000000001159
2. Siegert, S, Bahn, E, Kramer, ML, Schulz-Schaeffer, WJ, Hewett, JW, Breakefield, XO, et al. TorsinA expression is detectable in human infants as young as 4 weeks old. Brain Res Dev Brain Res (2005) 157(1):19–26. doi:10.1016/j.devbrainres.2005.02.019
3. Semple, BD, Blomgren, K, Gimlin, K, Ferriero, DM, and Noble-Haeusslein, LJ. Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol (2013) 106-107:1–16. doi:10.1016/j.pneurobio.2013.04.001
4. Vanni, V, Puglisi, F, Bonsi, P, Ponterio, G, Maltese, M, Pisani, A, et al. Cerebellar synaptogenesis is compromised in mouse models of DYT1 dystonia. Exp Neurol (2015) 271:457–67. doi:10.1016/j.expneurol.2015.07.005
5. Xiao, J, Gong, S, Zhao, Y, and LeDoux, MS. Developmental expression of rat torsinA transcript and protein. Brain Res Dev Brain Res (2004) 152(1):47–60. doi:10.1016/j.devbrainres.2004.05.012
6. Lohmann, K, and Klein, C. Genetics of dystonia: what’s known? What’s new? What’s next? Mov Disord (2013) 28(7):899–905. doi:10.1002/mds.25536
7. Bressman, SB, Sabatti, C, Raymond, D, de Leon, D, Klein, C, Kramer, PL, et al. The DYT1 phenotype and guidelines for diagnostic testing. Neurology (2000) 54(9):1746–52. doi:10.1212/wnl.54.9.1746
8. Ozelius, L, and Lubarr, N. DYT1 early-onset isolated dystonia. In: GeneReviews. Seattle: University of Washington (2016).
9. Zimmerman, CN, Jaunarajs, KLE, Meringolo, M, Rizzo, FR, Santoro, M, Standaert, DG, et al. Evaluation of AZD1446 as a therapeutic in DYT1 dystonia. Front Syst Neurosci (2017) 11:43. doi:10.3389/fnsys.2017.00043
10. Mazere, J, Dilharreguy, B, Catheline, G, Vidailhet, M, Deffains, M, Vimont, D, et al. Striatal and cerebellar vesicular acetylcholine transporter expression is disrupted in human DYT1 dystonia. Brain (2021) 144(3):909–23. doi:10.1093/brain/awaa465
11. Gras, C, Amilhon, B, Lepicard, EM, Poirel, O, Vinatier, J, Herbin, M, et al. The vesicular glutamate transporter VGLUT3 synergizes striatal acetylcholine tone. Nat Neurosci (2008) 11(3):292–300. doi:10.1038/nn2052
12. Higley, MJ, Gittis, AH, Oldenburg, IA, Balthasar, N, Seal, RP, Edwards, RH, et al. Cholinergic interneurons mediate fast VGluT3-dependent glutamatergic transmission in the striatum. PLoS One (2011) 6(4):e19155. doi:10.1371/journal.pone.0019155
13. Trudeau, L-E, and El Mestikawy, S. Glutamate cotransmission in cholinergic, GABAergic and monoamine systems: contrasts and commonalities. Front Neural Circuits (2018) 12:113. doi:10.3389/fncir.2018.00113
14. Mallet, N, Leblois, A, Maurice, N, and Beurrier, C. Striatal cholinergic interneurons: how to elucidate their function in health and disease. Front Pharmacol (2019) 10:1488. doi:10.3389/fphar.2019.01488
15. Lieberman, OJ, McGuirt, AF, Mosharov, EV, Pigulevskiy, I, Hobson, BD, Choi, S, et al. Dopamine triggers the maturation of striatal spiny projection neuron excitability during a critical period. Neuron (2018) 99(3):540–54. doi:10.1016/j.neuron.2018.06.044
16. Cai, Y, Nielsen, BE, Boxer, EE, Aoto, J, and Ford, CP. Loss of nigral excitation of cholinergic interneurons contributes to parkinsonian motor impairments. Neuron (2021) 109(7):1137–149.e5. doi:10.1016/j.neuron.2021.01.028
17. Braz, BY, Galiñanes, GL, Taravini, IR, Belforte, JE, and Murer, MG. Altered corticostriatal connectivity and exploration/exploitation imbalance emerge as intermediate phenotypes for a neonatal dopamine dysfunction. Neuropsychopharmacology (2015) 40:2576–87. doi:10.1038/npp.2015.104
18. Ztaou, S, and Amalric, M. Contribution of cholinergic interneurons to striatal pathophysiology in Parkinson’s disease. Neurochem Int (2019) 126:1–10. doi:10.1016/j.neuint.2019.02.019
19. Smith, R, Chung, H, Rundquist, S, Maat-Schieman, MLC, Colgan, L, Englund, E, et al. Cholinergic neuronal defect without cell loss in Huntington’s disease. Hum Mol Genet (2006) 15(21):3119–31. doi:10.1093/hmg/ddl252
20. Kataoka, Y, Kalanithi, PSA, Grantz, H, Schwartz, ML, Saper, C, Leckman, JF, et al. Decreased number of parvalbumin and cholinergic interneurons in the striatum of individuals with Tourette syndrome. J Comp Neurol (2010) 518(3):277–91. doi:10.1002/cne.22206
21. Bressman, SB, Hunt, AL, Heiman, GA, Brin, MF, Burke, RE, Fahn, S, et al. Exclusion of the DYT1 locus in a non-Jewish family with early-onset dystonia. Mov Disord (1994) 9(6):626–32. doi:10.1002/mds.870090608
22. Ozelius, L, Kramer, PL, Moskowitz, CB, Kwiatkowski, DJ, Brin, MF, Bressman, SB, et al. Human gene for torsion dystonia located on chromosome 9q32-q34. Neuron (1989) 2:1427–34. doi:10.1016/0896-6273(89)90188-8
23. Shashidharan, P, Sandu, D, Potla, U, Armata, IA, Walker, RH, McNaught, KS, et al. Transgenic mouse model of early-onset DYT1 dystonia. Hum Mol Genet (2005) 14(1):125–33. doi:10.1093/hmg/ddi012
24. Pappas, SS, Darr, K, Holley, SM, Cepeda, C, Mabrouk, OS, Wong, J-MT, et al. Forebrain deletion of the dystonia protein torsinA causes dystonic-like movements and loss of striatal cholinergic neurons. Elife (2015) 4:e08352. doi:10.7554/eLife.08352
25. Goodchild, RE, Kim, CE, and Dauer, WT. Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron (2005) 48(6):923–32. doi:10.1016/j.neuron.2005.11.010
26. Liang, C-C, Tanable, LM, Jou, S, Chi, F, and Dauer, WT. TorsinA hypofunction causes abnormal twisting movements and sensorimotor circuit neurodegeneration. J Clin Invest (2014) 124(7):3080–92. doi:10.1172/JCI72830
27. Weisheit, CE, and Dauer, WT. A novel conditional knock-in approach defines molecular and circuit effects of the DYT1 dystonia mutation. Hum Mol Genet (2015) 24(22):6459–72. doi:10.1093/hmg/ddv355
28. Maltese, M, Stanic, J, Tassone, A, Sciamanna, G, Ponterio, G, Vanni, V, et al. Early structural and functional plasticity alterations in a susceptibility period of DYT1 dystonia mouse striatum. Elife (2018) 7:e33331. doi:10.7554/eLife.33331
29. Sciamanna, G, Tassone, A, Martella, G, Mandolesi, G, Puglisi, F, Cuomo, D, et al. Developmental profile of the aberrant dopamine D2 receptor response in striatal cholinergic interneurons in DYT1 dystonia. PLoS One (2011) 6(9):e24261. doi:10.1371/journal.pone.0024261
30. Pappas, SS, Liang, C-C, Kim, S, Rivera, CO, and Dauer, WT. TorsinA dysfunction causes persistent neuronal nuclear pore defects. Hum Mol Genet (2018) 27(3):407–20. doi:10.1093/hmg/ddx405
31. Tanabe, LM, Liang, C-C, and Dauer, WT. Neuronal nuclear membrane budding occurs during a developmental window modulated by torsin paralogs. Cell Rep (2016) 16:3322–33. doi:10.1016/j.celrep.2016.08.044
32. Kim, S, Phan, S, Shaw, TR, Ellisman, MH, Veatch, SL, Barmada, SJ, et al. TorsinA is essential for the timing and localization of neuronal nuclear pore complex biogenesis. bioRxiv (2023).
33. Wiegert, JS, and Bading, H. Activity-dependent calcium signaling and ERK-MAP kinases in neurons: a link to structural plasticity of the nucleus and gene transcription regulation. Cell Calcium (2011) 49(5):296–305. doi:10.1016/j.ceca.2010.11.009
34. Granata, A, Koo, SJ, Haucke, V, Schiavo, G, and Warner, TT. CSN complex controls the stability of selected synaptic proteins via a torsinA-dependent process. EMBO J (2010) 30:181–93. doi:10.1038/emboj.2010.285
35. Rosenberg, SS, and Spitzer, NC. Calcium signaling in neuronal development. Cold Spring Harb Perspect Biol (2011) 3(10):a004259. doi:10.1101/cshperspect.a004259
36. Li, J, Levin, DS, Kim, AJ, Pappas, SS, and Dauer, WT. TorsinA restoration in a mouse model identifies a critical therapeutic window for DYT1 dystonia. J Clin Investig Investig (2021) 131(6):e139606. doi:10.1172/JCI139606
37. Yellajoshyula, D, Opeyemi, S, Dauer, WT, and Pappas, SS. Genetic evidence of aberrant striatal synaptic maturation and secretory pathway alteration in a dystonia mouse model. Dystonia (2022) 1:10892. doi:10.3389/dyst.2022.10892
38. Martella, G, Tassone, A, Sciamanna, G, Platania, P, Cuomo, D, Viscomi, MT, et al. Impairment of bidirectional synaptic plasticity in the striatum of a mouse model of DYT1 dystonia: role of endogenous acetylcholine. Brain (2009) 132(Pt 9):2336–49. doi:10.1093/brain/awp194
39. Shen, W, Flajolet, M, Greengard, P, and Surmeier, DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science (2008) 321(5890):848–51. doi:10.1126/science.1160575
40. Surmeier, DJ, Ding, J, Day, M, Wang, Z, and Shen, W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci (2007) 30(5):228–35. doi:10.1016/j.tins.2007.03.008
41. Balcioglu, A, Kim, M-O, Sharma, N, Cha, J-H, Breakefield, XO, and Standaert, DG. Dopamine release is impaired in a mouse model of DYT1 dystonia. J Neurochem (2007) 102(3):783–8. doi:10.1111/j.1471-4159.2007.04590.x
42. Xing, H, Yokoi, F, Walker, AL, Torres-Medina, R, Liu, Y, and Li, Y. Electrophysiological characterization of the striatal cholinergic interneurons in Dyt1 ΔGAG knock-in mice. Dystonia (2022) 1:10557. doi:10.3389/dyst.2022.10557
43. Yokoi, F, Oleas, J, Xing, H, Liu, Y, Dexter, KM, Misztal, C, et al. Decreased number of striatal cholinergic interneurons and motor deficits in dopamine receptor 2-expressing-cell-specific Dyt1 conditional knockout mice. Neurobiol Dis (2020) 134:104638. doi:10.1016/j.nbd.2019.104638
44. Pisani, A, Martella, G, Tscherter, A, Bonsi, P, Sharma, N, Bernardi, G, et al. Altered responses to dopaminergic D2 receptor activation and N-type calcium currents in striatal cholinergic interneurons in a mouse model of DYT1 dystonia. Neurobiol Dis (2006) 24(2):318–25. doi:10.1016/j.nbd.2006.07.006
45. Grundmann, K, Glöckle, N, Martella, G, Sciamanna, G, Hauser, T-K, Yu, L, et al. Generation of a novel rodent model for DYT1 dystonia. Neurobiol Dis (2012) 47(1):61–74. doi:10.1016/j.nbd.2012.03.024
46. Sciamanna, G, Hollis, R, Ball, C, Martella, G, Tassone, A, Marshall, A, et al. Cholinergic dysregulation produced by selective inactivation of the dystonia-associated protein torsinA. Neurobiol Dis (2012) 47(3):416–27. doi:10.1016/j.nbd.2012.04.015
47. Sciamanna, G, Tassone, A, Mandolesi, G, Puglisi, F, Ponterio, G, Martella, G, et al. Cholinergic dysfunction alters synaptic integration between thalamostriatal and corticostriatal inputs in DYT1 dystonia. J Neurosci (2012) 32(35):11991–2004. doi:10.1523/JNEUROSCI.0041-12.2012
48. Dang, MT, Yokoi, F, Cheetham, CC, Lu, J, Vo, V, Lovinger, DM, et al. An anticholinergic reverses motor control and corticostriatal LTD deficits in Dyt1 ΔGAG knock-in mice. Behav Brain Res (2012) 226(2):465–72. doi:10.1016/j.bbr.2011.10.002
49. Maltese, M, Martella, G, Madeo, G, Fagiolo, I, Tassone, A, Ponterio, G, et al. Anticholinergic drugs rescue synaptic plasticity in DYT1 dystonia: role of M1 muscarinic receptors. Mov Disord (2014) 29(13):1655–65. doi:10.1002/mds.26009
50. Gilbertson, T, Arkadir, D, and Steele, JD. Opposing patterns of abnormal D1 and D2 receptor dependent cortico-striatal plasticity explain increased risk taking in patients with DYT1 dystonia. PLoS One (2020) 15(5):e0226790. doi:10.1371/journal.pone.0226790
51. Arkadir, D, Radulescu, A, Raymond, D, Lubarr, N, Bressman, SB, Mazzoni, P, et al. DYT1 dystonia increases risk taking in humans. Elife (2016) 5:e14155. doi:10.7554/eLife.14155
52. Gemperli, K, Lu, X, Chintalapati, K, Rust, A, Bajpai, R, Suh, N, et al. Chronic striatal cholinergic interneuron excitation induces clinically-relevant dystonic behavior in mice. bioRxiv (2023).
53. Liu, Y, Xing, H, Sheng, W, Singh, KN, Korkmaz, AG, Comeau, C, et al. Alteration of the cholinergic system and motor deficits in cholinergic neuron-specific Dyt1 knockout mice. Neurobiol Dis (2021) 154:105342. doi:10.1016/j.nbd.2021.105342
54. Fox, WM. Reflex-ontogeny and behavioural development of the mouse. Anim Behav (1965) 13(2):234–41. doi:10.1016/0003-3472(65)90041-2
55. Flurkey, K, Currer, JM, and Harrison, DE. Mouse models in aging research. In: The mouse in biomedical research. 2nd ed. American College of Laboratory Animal Medicine (2007). p. 637–72.
56. Fishell, G, and van der Kooy, D. Pattern formation in the striatum: developmental changes in the distribution of striatonigral projections. Brain Res Dev Brain Res (1989) 45(2):239–55. doi:10.1016/0165-3806(89)90042-4
57. Jain, M, Armstrong, RJ, Barker, RA, and Rosser, AE. Cellular and molecular aspects of striatal development. Brain Res Bull (2001) 55(4):533–40. doi:10.1016/s0361-9230(01)00555-x
58. Basile, GA, Bertino, S, Bramanti, A, Ciurleo, R, Anastasi, GP, Milardi, D, et al. Striatal topographical organization: bridging the gap between molecules, connectivity and behavior. Eur J Histochem (2021) 65(s1):3284. doi:10.4081/ejh.2021.3284
59. Liao, W-L, Tsai, H-C, Wang, H-F, Chang, J, Lu, K-M, Wu, H-L, et al. Modular patterning of structure and function of the striatum by retinoid receptor signaling. Proc Natl Acad Sci USA (2008) 105(18):6765–70. doi:10.1073/pnas.0802109105
60. Knowles, R, Dehorter, N, and Ellender, T. From progenitors to progeny: shaping striatal circuit development and function. J Neurosci (2021) 41(46):9483–502. doi:10.1523/JNEUROSCI.0620-21.2021
61. Breakefield, XO, Blood, AJ, Li, Y, Hallett, M, Hanson, PI, and Standaert, DG. The pathophysiological basis of dystonias. Nat Rev Neurosci (2008) 9(3):222–34. doi:10.1038/nrn2337
62. de Carvalho Aguiar, PM, and Ozelius, LJ. Classification and genetics of dystonia. Lancet Neurol (2002) 1(5):316–25. doi:10.1016/s1474-4422(02)00137-0
63. Németh, AH. The genetics of primary dystonias and related disorders. Brain (2002) 125(4):695–721. doi:10.1093/brain/awf090
64. Schwarz, CS, and Bressman, SB. Genetics and treatment of dystonia. Neurol Clin (2009) 27(3):697–718. doi:10.1016/j.ncl.2009.04.010
65. Tanabe, LM, Kim, CE, Alagem, N, and Dauer, WT. Primary dystonia: molecules and mechanisms. Nat Rev Neurol (2009) 5(11):598–609. doi:10.1038/nrneurol.2009.160
66. Neychev, VK, Gross, RE, Lehéricy, S, Hess, EJ, and Jinnah, HA. The functional neuroanatomy of dystonia. Neurobiol Dis (2011) 42(2):185–201. doi:10.1016/j.nbd.2011.01.026
67. Zoons, E, Booij, J, Nederveen, AJ, Dijk, JM, and Tijssen, MAJ. Structural, functional and molecular imaging of the brain in primary focal dystonia—a review. Neuroimage. (2011) 56(3):1011–20. doi:10.1016/j.neuroimage.2011.02.045
68. Song, C-H, Bernhard, D, Hess, EJ, and Jinnah, HA. Subtle microstructural changes of the cerebellum in a knock-in mouse model of DYT1 dystonia. Neurobiol Dis (2014) 62:372–80. doi:10.1016/j.nbd.2013.10.003
69. Chen, L, Chatterjee, M, and Li, JYH. The mouse homeobox gene Gbx2 is required for the development of cholinergic interneurons in the striatum. J Neurosci (2010) 30(44):14824–34. doi:10.1523/JNEUROSCI.3742-10.2010
70. Poppi, LA, Ho-Nguyen, KT, Shi, A, Daut, CT, and Tischfield, MA. Recurrent implication of striatal cholinergic interneurons in a range of neurodevelopmental, neurodegenerative, and neuropsychiatric disorders. Cells (2021) 10(4):907. doi:10.3390/cells10040907
71. van Vulpen, EH, and van der Kooy, D. Striatal cholinergic interneurons: birthdates predict compartmental localization. Brain Res Dev Brain Res (1998) 109(1):51–8. doi:10.1016/s0165-3806(98)00012-1
72. Fentress, JC, Stanfield, BB, and Cowan, WM. Observations on the development of the striatum in mice and rats. Anat Embryol (Berl) (1981) 163(3):275–98. doi:10.1007/BF00315705
73. van Vulpen, EH, and Van Der Kooy, D. Differential maturation of cholinergic interneurons in the striatal patch versus matrix compartments. J Comp Neurol (1996) 365(4):683–91. doi:10.1002/(SICI)1096-9861(19960219)365:4<683::AID-CNE12>3.0.CO;2-I
74. Wright, J, Stanic, D, and Thompson, LH. Generation of striatal projection neurons extends into the neonatal period in the rat brain. J Physiol (2013) 591(1):67–76. doi:10.1113/jphysiol.2012.246397
75. Gould, E, Woolf, NJ, and Butcher, LL. Postnatal development of cholinergic neurons in the rat: I. Forebrain. Brain Res Bull (1991) 27(6):767–89. doi:10.1016/0361-9230(91)90209-3
76. Coyle, JT, and Yamamura, HI. Neurochemical aspects of the ontogenesis of cholinergic neurons in the rat brain. Brain Res (1976) 118:429–40. doi:10.1016/0006-8993(76)90310-3
77. Light, KE, Serbus, DC, and Santiago, M. Exposure of rats to ethanol from postnatal Days 4 to 8: alterations of cholinergic neurochemistry in the cerebral cortex and corpus striatum at day 20. Alcohol Clin Exp Res (1989) 13(1):29–35. doi:10.1111/j.1530-0277.1989.tb00279.x
78. Kuhar, MJ, Birdsall, NJM, Burgen, ASV, and Hulme, EC. Ontogeny of muscarinic receptors in the rat brain. Brain Res (1980) 184:375–83. doi:10.1016/0006-8993(80)90806-9
79. Kuo, H-Y, and Liu, F-C. Synaptic wiring of corticostriatal circuits in basal ganglia: insights into the pathogenesis of neuropsychiatric disorders. eNeuro (2019) 6(3):ENEURO.0076–19.2019. doi:10.1523/ENEURO.0076-19.2019
80. Tepper, JM, Sharpe, NA, Koós, TZ, and Trent, F. Postnatal development of the rat neostriatum: electrophysiological, light-and electron-microscopic studies. Dev Neurosci (1998) 20(2–3):125–45. doi:10.1159/000017308
81. McGuirt, AF, Post, MR, Pigulevskiy, I, Sulzer, D, and Lieberman, OJ. Coordinated postnatal maturation of striatal cholinergic interneurons and dopamine release dynamics in mice. J Neurosci (2021) 41(16):3597–609. doi:10.1523/JNEUROSCI.0755-20.2021
82. Sharpe, NA, and Tepper, JM. Postnatal development of excitatory synaptic input to the rat neostriatum: an electron microscopic study. Neuroscience (1998) 84(4):1163–75. doi:10.1016/s0306-4522(97)00583-6
83. Altman, J, and Sudarshan, K. Postnatal development of locomotion in the laboratory rat. Anim Behav (1975) 23(4):896–920. doi:10.1016/0003-3472(75)90114-1
84. Lee, H, Leamey, CA, and Sawatari, A. Perineuronal nets play a role in regulating striatal function in the mouse. PLoS One (2012) 7(3):e32747. doi:10.1371/journal.pone.0032747
85. Clarke, KA, and Still, J. Development and consistency of gait in the mouse. Physiol Behav (2001) 73(102):159–64. doi:10.1016/s0031-9384(01)00444-9
86. Shriner, AM, Drever, FR, and Metz, GA. The development of skilled walking in the rat. Behav Brain Res (2009) 205(2):426–35. doi:10.1016/j.bbr.2009.07.029
87. Abudukeyoumu, N, Hernandez-Flores, T, Garcia-Munoz, M, and Arbuthnott, GW. Cholinergic modulation of striatal microcircuits. Eur J Neurosci (2019) 49(5):604–22. doi:10.1111/ejn.13949
88. Doig, NM, Magill, PJ, Apicella, P, Bolam, JP, and Sharott, A. Cortical and thalamic excitation mediate the multiphasic responses of striatal cholinergic interneurons to motivationally salient stimuli. J Neurosci (2014) 34(8):3101–17. doi:10.1523/JNEUROSCI.4627-13.2014
89. Sizemore, RJ, Reynolds, JNJ, and Oorschot, DE. Number and type of synapses on the distal dendrite of a rat striatal cholinergic interneuron: a quantitative, ultrastructural study. J Anat (2010) 217(3):223–35. doi:10.1111/j.1469-7580.2010.01264.x
90. Goldberg, JA, and Reynolds, JNJ. Spontaneous firing and evoked pauses in the tonically active cholinergic interneurons of the striatum. Neuroscience (2011) 198:27–43. doi:10.1016/j.neuroscience.2011.08.067
91. Alcantara, AA, Chen, V, Herring, BE, Mendenhall, JM, and Berlanga, ML. Localization of dopamine D2 receptors on cholinergic interneurons of the dorsal striatum and nucleus accumbens of the rat. Brain Res (2003) 986(1–2):22–9. doi:10.1016/s0006-8993(03)03165-2
92. Bergson, C, Mrzljak, L, Smiley, JF, Pappy, M, Levenson, R, and Goldman-Rakic, PS. Regional, cellular, and subcellular variations in the distribution of D1 and D5 dopamine receptors in primate brain. J Neurosci (1995) 15(12):7821–36. doi:10.1523/JNEUROSCI.15-12-07821.1995
93. Yan, Z, and Surmeier, DJ. D5 dopamine receptors enhance Zn2+-sensitive GABAA currents in striatal cholinergic interneurons through a PKA/PP1 cascade. Neuron (1997) 19(5):1115–26. doi:10.1016/s0896-6273(00)80402-x
94. Yan, Z, Song, WJ, and Surmeier, J. D2 dopamine receptors reduce N-type Ca2+ currents in rat neostriatal cholinergic interneurons through a membrane-delimited, protein-kinase-C-insensitive pathway. J Neurophysiol (1997) 77(2):1003–15. doi:10.1152/jn.1997.77.2.1003
95. Oberlin, SR, Konakova, M, Pulst, S, and Chesselet, M-F. Development and anatomic localization of torsinA. Adv Neurol (2004) 94:61–5.
96. Hewett, JW, Tannous, B, Niland, BP, Nery, FC, Zeng, J, Li, Y, et al. Mutant torsinA interferes with protein processing through the secretory pathway in DYT1 dystonia cells. Proc Natl Acad Sci USA (2007) 104(17):7271–6. doi:10.1073/pnas.0701185104
97. Magno, L, Kretz, O, Bert, B, Ersözlü, S, Vogt, J, Fink, H, et al. The integrity of cholinergic basal forebrain neurons depends on expression of Nkx2-1. Eur J Neurosci (2011) 34(11):1767–82. doi:10.1111/j.1460-9568.2011.07890.x
98. Magno, L, Barry, C, Schmidt-Hieber, C, Theodotou, P, Häusser, M, and Kessaris, N. NKX2-1 is required in the embryonic septum for cholinergic system development, learning, and memory. Cell Rep (2017) 20(7):1572–84. doi:10.1016/j.celrep.2017.07.053
99. McGuirt, AF, Pigulevskiy, I, and Sulzer, DL. Developmental regulation of thalamus-driven pauses in striatal cholinergic interneurons. iScience (2022) 25:105332. doi:10.1016/j.isci.2022.105332
100. Oz, O, Matityahu, L, Mizrahi-Kliger, A, Kaplan, A, Berkowitz, N, Tiroshi, L, et al. Non-uniform distribution of dendritic nonlinearities differentially engages thalamostriatal and corticostriatal inputs onto cholinergic interneurons. Elife (2022) 11:e76039. doi:10.7554/eLife.76039
101. Zhang, Y-F, Reynolds, JNJ, and Cragg, SJ. Pauses in cholinergic interneuron activity are driven by excitatory input and delayed rectification, with dopamine modulation. Neuron (2018) 98(5):918–25. doi:10.1016/j.neuron.2018.04.027
102. Carrillo-Reid, L, Tecuapetla, F, Ibáñez-Sandoval, O, Hernández-Cruz, A, Elvira Galarraga, A, and Bargas, J. Activation of the cholinergic system endows compositional properties to striatal cell assemblies. J Neurophysiol (2009) 101(2):737–49. doi:10.1152/jn.90975.2008
103. Silberstein, P, Kühn, AA, Kupsch, A, Trottenberg, T, Krauss, JK, Wöhrle, JC, et al. Patterning of globus pallidus local field potentials differs between Parkinson’s disease and dystonia. Brain (2003) 126(Pt 12):2597–608. doi:10.1093/brain/awg267
104. Hendrix, CM, and Vitek, JL. Pathophysiology of dystonia: models and mechanisms. In: Dystonia and dystonia syndromes. Vienna: Springer-Verlag (2015). p. 187–207.
105. Raz, A, Feingold, A, Zelanskaya, V, Vaadia, E, and Bergman, H. Neuronal synchronization of tonically active neurons in the striatum of normal and parkinsonian primates. J Neurophysiol (1996) 76(3):2083–8. doi:10.1152/jn.1996.76.3.2083
106. Magariños-Ascone, CM, Regidor, I, Gómez-Galán, M, Cabañes-Martínez, L, and Figueiras-Méndez, R. Deep brain stimulation in the globus pallidus to treat dystonia: electrophysiological characteristics and 2 years’ follow-up in 10 patients. Neuroscience (2008) 152(2):558–71. doi:10.1016/j.neuroscience.2008.01.001
107. Hahn, PJ, Russo, GS, Hashimoto, T, Miocinovic, S, Xu, W, McIntyre, CC, et al. Pallidal burst activity during therapeutic deep brain stimulation. Exp Neurol (2009) 211(1):243–51. doi:10.1016/j.expneurol.2008.01.032
108. Kondabolu, K, Roberts, EA, Bucklin, M, McCarthy, MM, Kopell, N, and Han, X. Striatal cholinergic interneurons generate beta and gamma oscillations in the corticostriatal circuit and produce motor deficits. Proc Natl Acad Sci USA (2016) 113(22):E3159–68. doi:10.1073/pnas.1605658113
109. Liu, X, Wang, S, Yianni, J, Nandi, D, Bain, PG, Gregory, R, et al. The sensory and motor representation of synchronized oscillations in the globus pallidus in patients with primary dystonia. Brain (2008) 131(Pt 6):1562–73. doi:10.1093/brain/awn083
110. Scarduzio, M, Zimmerman, CN, Jaunarajs, KL, Wang, Q, Standaert, DG, and McMahon, LL. Strength of cholinergic tone dictates the polarity of dopamine D2 receptor modulation of striatal cholinergic interneuron excitability in DYT1 dystonia. Exp Neurol (2017) 295:162–75. doi:10.1016/j.expneurol.2017.06.005
111. Hashimoto, M, and Mikoshiba, K. Mediolateral compartmentalization of the cerebellum is determined on the “birth date” of Purkinje cells. J Neurosci (2003) 23(36):11342–51. doi:10.1523/JNEUROSCI.23-36-11342.2003
112. Wallace, VA. Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol (1999) 9(8):445–8. doi:10.1016/s0960-9822(99)80195-x
113. Lewis, PM, Gritli-Linde, A, Smeyne, R, Kottmann, A, and McMahon, AP. Sonic hedgehog signaling is required for expansion of granule neuron precursors and patterning of the mouse cerebellum. Dev Biol (2004) 270(2):393–410. doi:10.1016/j.ydbio.2004.03.007
114. van der Heijden, ME, and Sillitoe, RV. Interactions between purkinje cells and granule cells coordinate the development of functional cerebellar circuits. Neuroscience (2021) 462:4–21. doi:10.1016/j.neuroscience.2020.06.010
115. van der Heijden, ME, Lackey, EP, Perez, R, Ișleyen, FS, Brown, AM, Donofrio, SG, et al. Maturation of Purkinje cell firing properties relies on neurogenesis of excitatory neurons. Elife (2021) 10:e68045. doi:10.7554/eLife.68045
116. Baader, SL, Sanlioglu, S, Berrebi, AS, Parker-Thornburg, J, and Oberdick, J. Ectopic overexpression of engrailed-2 in cerebellar Purkinje cells causes restricted cell loss and retarded external germinal layer development at lobule junctions. J Neurosci (1998) 18(5):1763–73. doi:10.1523/JNEUROSCI.18-05-01763.1998
117. Ohmori, H, Ogura, H, Yasuda, M, Nakamura, S, Hatta, T, Kawano, K, et al. Developmental neurotoxicity of phenytoin on granule cells and Purkinje cells in mouse cerebellum. J Neurochem (1999) 72(4):1497–506. doi:10.1046/j.1471-4159.1999.721497.x
118. Prastiwi, D, Djunaidi, A, and Partadiredja, G. High dosage of monosodium glutamate causes deficits of the motor coordination and the number of cerebellar Purkinje cells of rats. Hum Exp Toxicol (2015) 34(11):1171–9. doi:10.1177/0960327115572706
119. Pakhotin, P, and Bracci, E. Cholinergic interneurons control the excitatory input to the striatum. J Neurosci (2007) 27(2):391–400. doi:10.1523/JNEUROSCI.3709-06.2007
120. Kosillo, P, Zhang, Y-F, Threlfell, S, and Cragg, SJ. Cortical control of striatal dopamine transmission via striatal cholinergic interneurons. Cereb Cortex (2016) 26(11):4160–9. doi:10.1093/cercor/bhw252
121. Sreenivasan, V, Serafeimidou-Pouliou, E, Exposito-Alonso, D, Bercsenyi, K, Bernard, C, Bae, S-E, et al. Input-specific control of interneuron numbers in nascent striatal networks. Proc Natl Acad Sci U S A. (2022) 119(20):e2118430119. doi:10.1073/pnas.2118430119
122. Luis, COS, Sanchez-Garcia, MA, Nieto-Gonzalez, JL, García-Junco-Clemente, P, Montero-Sanchez, A, Fernandez-Chacon, R, et al. Substantia nigra dopaminergic neurons and striatal interneurons are engaged in three parallel but interdependent postnatal neurotrophic circuits. Aging Cell (2018) 17(5):e12821. doi:10.1111/acel.12821
123. Sanchez-Ortiz, E, Yui, D, Song, D, Li, Y, Rubenstein, JL, Reichardt, LF, et al. TrkA gene ablation in basal forebrain results in dysfunction of the cholinergic circuitry. J Neurosci (2012) 32(12):4065–79. doi:10.1523/JNEUROSCI.6314-11.2012
124. Opal, P, Tintner, R, Jankovic, J, Leung, J, Breakefield, XO, Friedman, J, et al. Intrafamilial phenotypic variability of the DYT1 dystonia: from asymptomatic TOR1A gene carrier status to dystonic storm. Mov Disord (2002) 17(2):339–45. doi:10.1002/mds.10096
125. Rauschenberger, L, Knorr, S, Pisani, A, Hallett, M, Volkmann, J, and Ip, CW. Second hit hypothesis in dystonia: dysfunctional cross talk between neuroplasticity and environment? Neurobiol Dis (2021) 159:105511. doi:10.1016/j.nbd.2021.105511
126. Partridge, JG, Tang, KC, and Lovinger, DM. Regional and postnatal heterogeneity of activity-dependent long-term changes in synaptic efficacy in the dorsal striatum. J Neurophysiol (2000) 84(3):1422–9. doi:10.1152/jn.2000.84.3.1422
127. Robbe, D. To move or to sense? Incorporating somatosensory representation into striatal functions. Curr Opin Neurobiol (2018) 52:123–30. doi:10.1016/j.conb.2018.04.009
128. Martella, G, Maltese, M, Nisticò, R, Schirinizi, T, Madeo, G, Sciamanna, G, et al. Regional specificity of synaptic plasticity deficits in a knock-in mouse model of DYT1 dystonia. Neurobiol Dis (2014) 65:124–32. doi:10.1016/j.nbd.2014.01.016
129. Tassone, A, Martella, G, Meringolo, M, Vanni, V, Sciamanna, G, Ponterio, G, et al. Vesicular acetylcholine transporter alters cholinergic tone and synaptic plasticity in DYT1 dystonia. Mov Disord (2021) 36(12):2768–79. doi:10.1002/mds.28698
130. Lemos, JC, Shin, JH, and Alvarez, VA. Striatal cholinergic interneurons are a novel target of corticotropin releasing factor. J Neurosci (2019) 39(29):5647–61. doi:10.1523/JNEUROSCI.0479-19.2019
131. Schulz, JM, and Reynolds, JNJ. Pause and rebound: sensory control of cholinergic signaling in the striatum. Trends Neurosci (2013) 36(1):41–50. doi:10.1016/j.tins.2012.09.006
132. Nava, E, and Röder, B. Adaptation and maladaptation: insights from brain plasticity. Progess Brain Res (2011) 191:177–94. doi:10.1016/B978-0-444-53752-2.00005-9
133. Li, J, Liang, C-C, Pappas, SS, and Dauer, WT. TorsinB overexpression prevents abnormal twisting in DYT1 dystonia mouse models. Elife (2020) 9:e54285. doi:10.7554/eLife.54285
134. Ozelius, LJ, Page, CE, Klein, C, Hewett, JW, Mineta, M, Leung, J, et al. The TOR1A (DYT1) gene family and its role in early onset torsion dystonia. Genomics (1999) 62(3):377–84. doi:10.1006/geno.1999.6039
135. Laudermilch, E, Tsai, P-L, Graham, M, Turner, E, Zhao, C, and Schlieker, C. Dissecting Torsin/cofactor function at the nuclear envelope: a genetic study. Mol Biol Cell (2016) 27(25):3964–71. doi:10.1091/mbc.E16-07-0511
136. Vasudevan, A, Breakefield, XO, and Bhide, PG. Developmental patterns of torsinA and torsinB expression. Comp Study (2006) 1073–1074:139–45. doi:10.1016/j.brainres.2005.12.087
137. Richter, F, Bauer, A, Perl, S, Schulz, A, and Richter, A. Optogenetic augmentation of the hypercholinergic endophenotype in DYT1 knock-in mice induced erratic hyperactive movements but not dystonia. EbioMedicine (2019) 41:649–58. doi:10.1016/j.ebiom.2019.02.042
138. Schulz, A, Richter, F, and Richter, A. In vivo optogenetic inhibition of striatal parvalbumin-reactive interneurons induced genotype-specific changes in neuronal activity without dystonic signs in male DYT1 knock-in mice. J Neurosci Res (2023) 101(4):448–63. doi:10.1002/jnr.25157
139. Yokoi, F, Chen, H-X, Dang, MT, Cheetham, CC, Campbell, SL, Roper, SN, et al. Behavioral and electrophysiological characterization of Dyt1 heterozygous knockout mice. PLoS One (2015) 10(3):e0120916. doi:10.1371/journal.pone.0120916
140. Demircioglu, FE, Sosa, BA, Ingram, J, Ploegh, HL, and Schwartz, TU. Structures of torsinA and its disease-mutant complexed with an activator reveal the molecular basis for primary dystonia. Elife (2016) 5:e17983. doi:10.7554/eLife.17983
141. Giles, LM, Chen, J, Li, L, and Chin, L-S. Dystonia-associated mutations cause premature degradation of torsinA protein and cell-type-specific mislocalization to the nuclear envelope. Hum Mol Genet (2008) 17(17):2712–22. doi:10.1093/hmg/ddn173
142. Torres, GE, Sweeney, AL, Beaulieu, J-M, Shashidharan, P, and Caron, MG. Effect of torsinA on membrane proteins reveals a loss of function and a dominant-negative phenotype of the dystonia-associated DeltaE-torsinA mutant. Proc Natl Acad Sci USA (2004) 101(44):15650–5. doi:10.1073/pnas.0308088101
143. Zhao, C, Brown, RSH, Chase, AR, Eisele, MR, and Schlieker, C. Regulation of torsin ATPases by LAP1 and LULL1. Proc Natl Acad Sci USA (2013) 110(17):E1545–54. doi:10.1073/pnas.1300676110
144. Pham, P, Frei, KP, Woo, W, and Truong, DD. Molecular defects of the dystonia-causing torsinA mutation. Neuroreport (2006) 17(16):1725–8. doi:10.1097/WNR.0b013e3280101220
145. Cruz, L, György, B, Cheah, PS, Kleinstiver, BP, Eimer, WA, Garcia, SP, et al. Mutant allele-specific CRISPR disruption in DYT1 dystonia fibroblasts restores cell function. Mol Ther Nucleic Acids (2020) 21:1–12. doi:10.1016/j.omtn.2020.05.009
146. Ding, B, Tang, Y, Ma, S, Akter, M, Liu, M-L, Zang, T, et al. Disease modeling with human neurons reveals LMNB1 dysregulation underlying DYT1 dystonia. J Neurosci (2021) 41(9):2024–38. doi:10.1523/JNEUROSCI.2507-20.2020
Keywords: development, DYT1, striatum, early-onset dystonia, cholinergic interneurons
Citation: Miterko-Myers LN (2024) Striatal cholinergic interneuron development in models of DYT1 dystonia. Dystonia 3:12413. doi: 10.3389/dyst.2024.12413
Received: 14 November 2023; Accepted: 02 April 2024;
Published: 03 May 2024.
Edited by:
Aasef Shaikh, Case Western Reserve University, United StatesCopyright © 2024 Miterko-Myers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lauren N. Miterko-Myers, bGF1cmVuLm1pdGVya29AdXRzb3V0aHdlc3Rlcm4uZWR1