 Hong Xing
Hong Xing Pallavi Girdhar1,2
Pallavi Girdhar1,2 Fumiaki Yokoi
Fumiaki Yokoi Yuqing Li
Yuqing Li- 1Norman Fixel Institute of Neurological Diseases, McKnight Brain Institute, University of Florida, Gainesville, FL, United States
- 2Department of Neurology, College of Medicine, University of Florida, Gainesville, FL, United States
Myoclonus is a hyperkinetic movement disorder characterized by sudden, brief, involuntary jerks of single or multiple muscles. Dystonia is a movement disorder characterized by sustained or intermittent muscle contractions causing abnormal, often repetitive, movements, postures, or both. Myoclonus-dystonia (M-D) or DYT11 dystonia is an early-onset genetic disorder characterized by subcortical myoclonus and less pronounced dystonia. DYT11 dystonia is the primary genetic M-D caused by loss of function mutations in SGCE, which codes for ε-sarcoglycan. Sgce knockout (KO) mice model DYT11 dystonia and exhibit myoclonus, motor deficits, and psychiatric-like behaviors. Neuroimaging studies show abnormal cerebellar activity in DYT11 dystonia patients. Acute small hairpin RNA (shRNA) knockdown of Sgce mRNA in the adult cerebellum leads to motor deficits, myoclonic-like jerky movements, and altered Purkinje cell firing. Whether Sgce KO mice show similar abnormal Purkinje cell firing as the acute shRNA knockdown mice is unknown. We used acute cerebellar slice recording in Sgce KO mice to address this issue. The Purkinje cells from Sgce KO mice showed spontaneous and intrinsic excitability changes compared to the wild-type (WT) mice. Intrinsic membrane properties were not altered. The female Sgce KO mice had more profound alterations in Purkinje cell firing than males, which may correspond to the early onset of the symptoms in female human patients and more pronounced myoclonus in female KO mice. Our results suggest that the abnormal Purkinje cell firing in the Sgce KO mice contributes to the manifestation of the myoclonus and other motor symptoms in DYT11 dystonia patients.
Introduction
DYT11 dystonia is a major type of genetic M-D and is caused by mutations in SGCE, which codes for ε-sarcoglycan [1]. Many mutations in SGCE, such as nonsense, missense, and frameshift mutations, have been reported in M-D patients, suggesting that the loss of ε-sarcoglycan function causes DYT11 dystonia [2, 3]. The primary symptom of DYT11 dystonia is myoclonus; however, dystonia and psychiatric symptoms, such as depression, panic, and obsessive-compulsive disorder, have been reported in some patients [1, 4]. Interestingly, alcohol consumption in some patients can provide temporal relief of the symptoms [3, 5]. Treatment has been focused on symptom relief using various drugs and deep brain stimulation with limited success [2, 6]. Female individuals outnumber male individuals for both adult-onset idiopathic and early-onset monogenic dystonias [7]. The average age of onset for female DYT11 patients is 5 years versus 8 years for male patients [8].
Animal models are helpful to investigate the pathophysiology of genetic diseases and contribute to developing better treatments. Multiple mouse lines have been generated and characterized to model DYT11 dystonia [9–15]. Sgce is the mouse homolog of the human SGCE gene. Sgce is maternally imprinted and paternally expressed [11, 16, 17]. We generated paternally-inherited Sgce heterozygous KO mice lacking exon four and analyzed their behavioral and neurochemical phenotypes [11, 15, 18]. The Sgce KO mice, on average, exhibit 28 times more myoclonus than WT littermates and have deficits in motor learning, anxiety, depression-like behaviors, and fine motor coordination and balance. Furthermore, we found that the striatal dopaminergic system is impaired in the Sgce KO mice. The levels of dopamine and its metabolites 3,4-dihydroxyphenylacetic acid (DOPAC) and 3-methoxy-4-hydroxyphenylacetic acid (homovanillic acid; HVA) in the striatum of Sgce KO mice are significantly higher than those of wild-type (WT) mice [15]. Sgce KO mice exhibit a significantly low level of striatal dopamine receptor 2 (D2R) and a significant increase in dopamine release after amphetamine injection in comparison to WT littermates [18]. Finally, striatal medium spiny neurons and cerebellar Purkinje cells show abnormal nuclear envelopes in Sgce KO mice [9, 10].
Cerebellar circuits, especially Purkinje cells, are central players in movement and posture control, and there are multiple lines of investigations implicating their involvement in DYT11 dystonia pathogenesis [19–24]. Several brain imaging studies show the involvement of the cerebellum in DYT11 patients [25–32]. DYT11 patients possess abnormal responses to cerebellar eye-blink classic conditioning [33, 34], which can be normalized by alcohol intake. Furthermore, DYT11 patients perform abnormally in a cerebellar saccadic adaptation task [35] but not in a limb adaptation task involving symptomatic body regions [36]. However, Purkinje cell-specific Sgce KO mice exhibit motor learning deficits but no myoclonus [10]. This suggests that Sgce KO in other cells inside and outside the cerebellum is needed to produce the myoclonus in mice. Interestingly, small hairpin RNA (shRNA) knockdown of Sgce mRNA in the adult cerebellum, but not basal ganglia, leads to motor deficits, spinning, and myoclonic-like jerky movements, which can be reduced by alcohol consumption. Moreover, the awake, head-restrained shRNA-treated mice show aberrant firing of Purkinje cells and deep cerebellar nuclei neurons in vivo [37]. The abnormal function of cerebellar circuits is likely involved in the pathogenesis of DYT11 dystonia. However, whether the Sgce KO mice, which have the Sgce gene inactivated constitutively throughout the lifespan, show similar aberrant Purkinje cell firing is unknown. Here, the Purkinje cells in the Sgce KO mice were characterized by electrophysiological recording of acute brain slices. The spontaneous firing, intrinsic excitability, and membrane properties of Purkinje cells were examined and compared to their myoclonus behavior published earlier [15].
Materials and methods
Animals
All experiments complied with the United States Public Health Service Guide for Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committees of the University of Florida. As described previously, Sgce KO mice and their WT littermates were prepared and genotyped by PCR [15]. Mice were housed under a 12-hour light and 12-hour dark cycle with ad libitum access to food and water. All experiments and initial data analysis were performed by investigators blind to the genotypes. This study followed the recommended heterogenization of study samples of various ages, and the data were analyzed with age as a covariate [38].
Brain slice electrophysiology
We used male and female mice to investigate the effects of sex on the phenotype. As described previously [39–43], electrophysiological recordings data for spontaneous firing, intrinsic excitability, and membrane properties of 105 Purkinje cells were obtained from 10 WT mice (5 males and 5 females) and 6 Sgce KO littermates (3 males and 3 females) at 80–165 days old. Briefly, the cell-attached recordings of Purkinje cells were performed in the parasagittal 300 μm-thick cerebellar brain slices. After recording the spontaneous firing, whole-cell recordings were made by breaking through the membrane. Tonic Purkinje cells fire relatively constantly, while non-tonic Purkinje cells fire intermittently, with pauses separating the firing periods [44]. We define tonic Purkinje cells as those cells without pause of the spontaneous firing lasting for more than 300 msec during the cell-attached recording [45] and otherwise as non-tonic cells [40]. The intrinsic properties (resting membrane potential, capacitance, membrane resistance, and time constant) were measured in the whole-cell recording mode. Finally, current steps were injected to determine intrinsic excitability.
Myoclonus
The myoclonus data from 17 KO (9 males and 8 females) and 22 WT littermates (11 males and 11 females), 210–241 days old, were published previously [15]. The data were reanalyzed here to determine the effect of sex on the number of myoclonus.
Statistics
Data were tested for normality first using the univariate procedure of the SAS statistical package. A generalized linear model (GENMOD) was used to compare the spontaneous firing, intrinsic excitability, membrane properties, and myoclonus. Age and body weight were used as continuous variables, and data from each cell were nested within individual animals and treated as repeated measurements. A negative binomial distribution was used for count data, i.e., the number of action potentials in the current injection and myoclonus. A gamma distribution was used for data that was not normally distributed. For tonic/non-tonic cell distribution analysis, chi-square was used. Significance was assigned at p < 0.05. A p-value between 0.05 and 0.1 was considered a trend. Data in the text are presented as “mean ± standard error of the mean (SEM)” unless specified otherwise.
Results
Altered spontaneous firing frequency and coefficient of variation (CV) of the Purkinje cells in Sgce KO mice
Cerebellar Purkinje cells are the sole output of the cerebellar cortex, projecting into the deep cerebellar nuclei, which are the sole output of the cerebellum. They play an essential role in cerebellar function. The Purkinje cells in the Sgce KO mice were characterized by acute brain slice recording to understand their role in the pathogenesis of DYT11 dystonia. The spontaneous firing of the Purkinje cells (WT, 59 cells/10 mice; KO, 45 cells/6 mice) was recorded by cell-attached recording mode with a voltage clamp. The representative traces of the Purkinje cells are shown in Figure 1A. Although the firing frequency (WT, 63.8 ± 7.7 Hz; KO, 53.8 ± 3.6; p = 0.22, Figure 1B) was not altered, the CV (WT, 0.338 ± 0.026; KO, 0.249 ± 0.013; p = 0.0009; Figure 1C) was significantly decreased in Sgce KO mice compared to WT mice. CV is an indication of firing regularity. A decreased CV suggests increased firing regularity. Purkinje cells can be grouped into tonic and non-tonic types [40, 43, 45]. When analyzed separately by the cell types, tonic cells showed no change in the frequency (WT, 35 cells/10 mice; 50.1 ± 5.5 Hz; KO, 22 cells/6 mice, 62.3 ± 10.2; p = 0.27, Figure 1B) but a significant decrease in CV (WT, 0.110 ± 0.005; KO, 0.090 ± 0.007; p = 0.026; Figure 1C), while non-tonic cells showed decreases both in the frequency (WT, 24 cells/10 mice, 81.5 ± 13.4 Hz; KO, 23 cells/6 mice, 48.3 ± 10.7; p = 0.059, Figure 1B) and CV (WT, 1.071 ± 0.151; KO, 0.720 ± 0.118; p = 0.061; Figure 1C). When separated by sex, female Purkinje cells showed a significant decrease in the frequency (WT, 27 cells/5 mice, 67.2 ± 7.5 Hz; KO, 20 cells/3 mice, 48.6 ± 5.2; p = 0.036, Figure 1B) but no change in CV (WT, 0.330 ± 0.024; KO, 0.295 ± 0.037; p = 0.43; Figure 1C), while male Purkinje cells showed no change in the frequency (WT, 32 cells/5 mice, 59.8 ± 13.4 Hz; KO, 25 cells/3 mice, 61.7 ± 7.0; p = 0.90, Figure 1B) but a significant decrease of CV (WT, 0.344 ± 0.033; KO, 0.212 ± 0.010; p < 0.001; Figure 1C). When separated by both sex and cell type, there were significant decreases in frequency of female non-tonic Purkinje cells (WT, 12 cells/5 mice, 80.0 ± 11.3 Hz; KO, 13 cells/3 mice, 40.1 ± 10.5; p = 0.02, Figure 1B) and in CV of male non-tonic Purkinje cells (WT, 12 cells/5 mice 1.232 ± 0.244; KO, 10 cells/3 mice, 0.534 ± 0.081; p = 0.0008; Figure 1C), while the frequency and CV of the rest remained unchanged (Figures 1B, C).
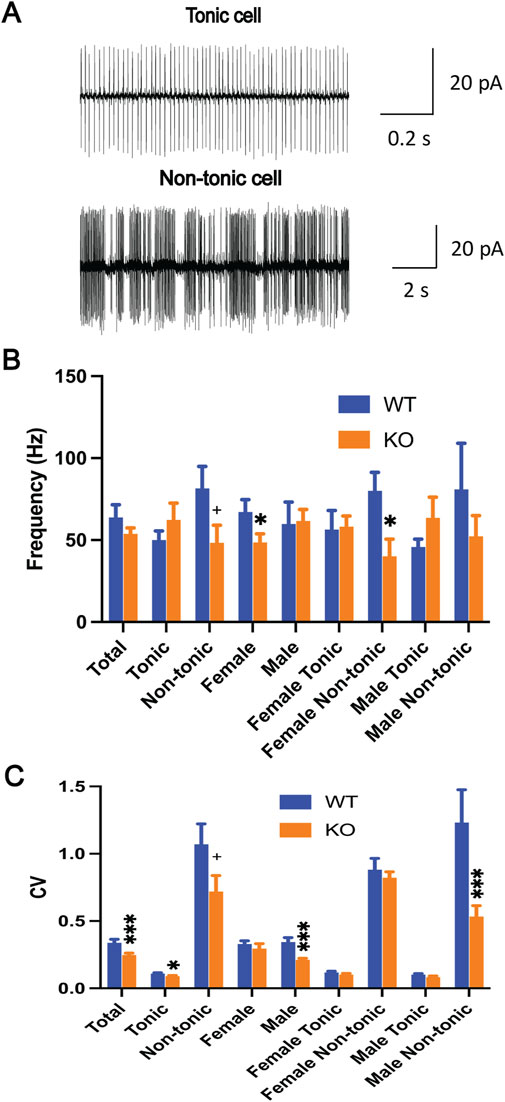
Figure 1. Spontaneous firing of the Purkinje cells in brain slices. (A) The representative traces of both the tonic and non-tonic Purkinje cells. Spontaneous firing frequency (B) and CV (C) were significantly altered in Sgce KO mice in a sex- and cell-type-specific manner (SAS GENMOD was used for statistical analysis). The bars represent means ± SEM. *, p < 0.05; ***, p < 0.001; + in (B), p = 0.059, in (C), p = 0.061.
Finally, the relative ratio of the tonic and non-tonic cells was analyzed, and there was no significant difference between the WT and Sgce KO mice (WT: tonic = 35, non-tonic = 24; KO: tonic = 22, non-tonic = 23, p = 0.29). Overall, Sgce KO mice had normal cell type distribution and sex-specific alteration of spontaneous firing of Purkinje cells both in frequency and CV, especially in the non-tonic cell types.
Altered intrinsic excitability of the Purkinje cells in Sgce KO mice
After recording the spontaneous firing by cell-attached mode, the intrinsic membrane properties were measured in whole-cell recording mode. The resting membrane property of the Purkinje cells was determined from 10 WT (56 cells) and 6 Sgce KO mice (46 cells). There was no significant difference in the membrane capacitance, membrane resistance, and resting membrane potential (RMP) between the WT and Sgce KO mice (Table 1). However, the Sgce KO mice showed increased membrane constants compared to the WT mice (p = 0.053), largely derived from females (p = 0.054). Male and female mice did not differ in the other 3 parameters (Table 1).
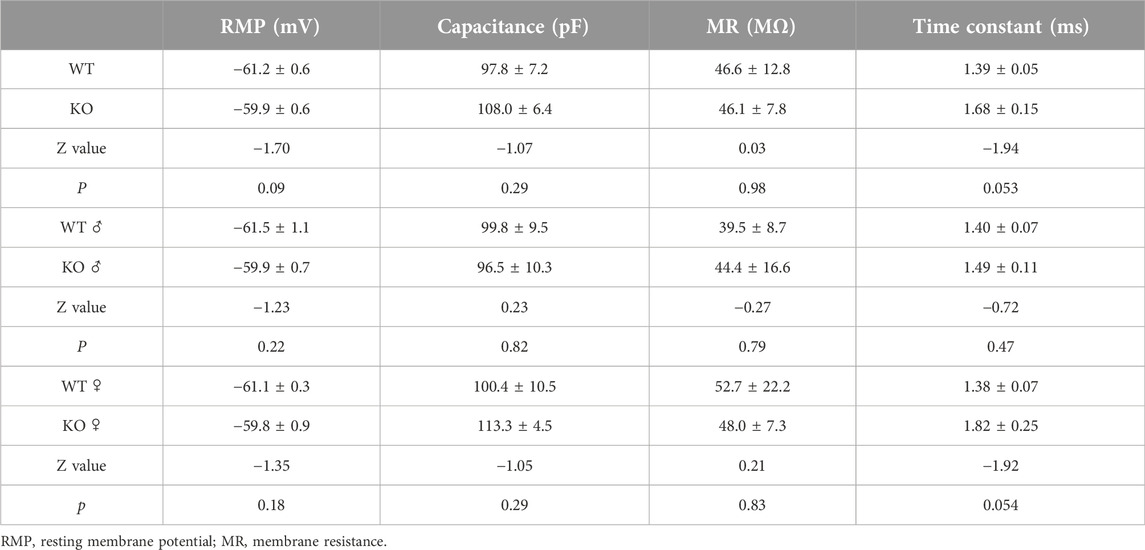
Table 1. Intrinsic properties of Purkinje cells.
The intrinsic excitability of the Purkinje cells in the brain slices was measured with current step injections. The recorded neurons showed typical electrophysiological responses of the Purkinje cells (Figure 2A). The number of action potentials fired overall (WT, 56 cells/10 mice, 32.9 ± 2.1; KO, 46 cells/6 mice, 31.7 ± 1.8; p = 0.67, Figure 2B) and at each current step (Figure 2C) were similar between WT and Sgce KO mice. When analyzed separately by sex, female Purkinje cells showed a significant decrease in the number of action potentials (WT, 30 cells/5 mice, 36.9 ± 3.6; KO, 24 cells/3 mice, 27.8 ± 1.4; p = 0.0096, Figure 2B), while male Purkinje cells showed a significant increase (WT, 26 cells/5 mice, 28.8 ± 1.2; KO, 22 cells/3 mice, 36.2 ± 2.2; p = 0.0019, Figure 2B). When separated by both sex and current steps, there were significant decreases in the number of action potentials from steps 2 to 6 in the female KO mice compared to the WT mice (Figure 2D). In contrast, the male KO mice had a significant increase at the 8th step and a potential increase at the 2nd step (Figure 2E). These results suggest that the intrinsic excitability of the Purkinje cells is altered in the Sgce KO mice in a sex-specific manner.
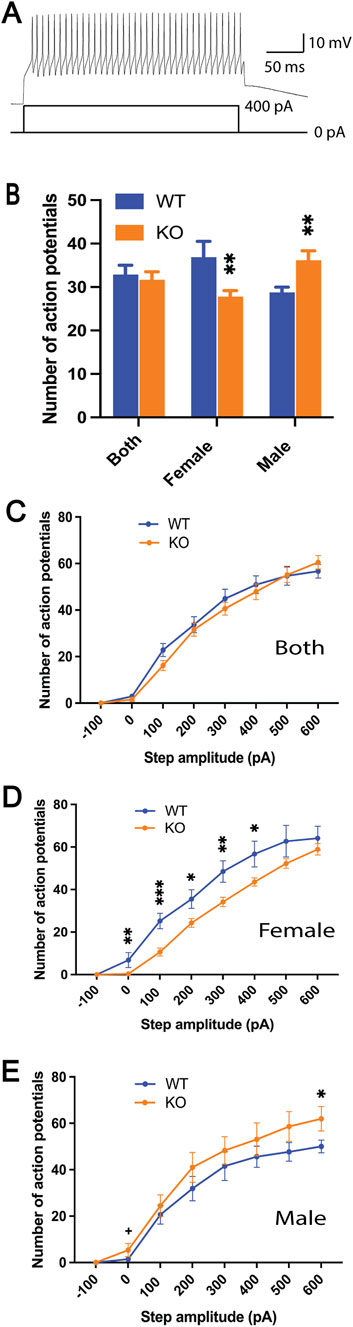
Figure 2. Intrinsic excitability of Purkinje cells as measured by current steps in the brain slices. Representative trace of the action potential firing in response to the current injection (A). The number of action potentials of all eight steps combined (B) and at each current step (C–E) were significantly altered in Sgce KO mice in a sex-specific manner (SAS GENMOD was used for statistical analysis). Means ± SEM were plotted. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Increased myoclonus in Sgce KO mice and sex difference
We previously reported that Sgce KO mice, on average, exhibit 28 times more myoclonus than WT littermates [15]. Here, we reanalyzed the data separately by each sex or genotype. Both male and female Sgce KO mice showed significantly increased numbers of myoclonus compared to the WT mice (Females: WT, n = 11, 0.8 ± 0.5; KO, n = 8, 43.4 ± 26.3; p = <0.0001; Males: WT, n = 11, 0.5 ± 0.4; KO, n = 9, 9.4 ± 6.3; p = 0.014, Figure 3). While WT males and females showed no difference (p = 0.29), the KO females showed four times more myoclonus compared to the KO males (p = 0.0048), suggesting female KO mice have a more pronounced myoclonus phenotype compared to male KO mice.
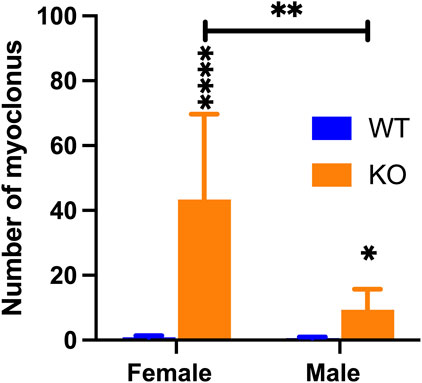
Figure 3. Comparison of the numbers of myoclonus in male and female WT and Sgce KO mice. While WT males and females did not differ, female Sgce KO mice showed 4 times more myoclonus than male Sgce KO mice (SAS GENMOD was used for statistical analysis). Means ± SEM were plotted. *, p < 0.05; **, p < 0.01; ****, p < 0.0001.
Discussion
We aimed to determine whether there was abnormal Purkinje cell firing in the Sgce KO mice, a model for DYT11 dystonia or M-D. For spontaneous firing, the Sgce KO mice showed a significantly decreased CV and no change in the frequency. Interestingly, the female Sgce KO mice showed significantly decreased frequency, especially in non-tonic cells, while male Sgce KO mice had significantly decreased CV, derived mainly from non-tonic cells. For intrinsic excitability, female Sgce KO mice showed a significant decrease, consistent with their decreased spontaneous firing frequency. In contrast, male Sgce KO mice showed a modest increase that did not lead to increased spontaneous firing. Membrane properties remained unchanged except for the membrane constant, which showed an increasing trend in females. Female Sgce KO mice showed profound electrophysiological changes in the Purkinje cells compared to male Sgce KO mice. To correlate myoclonus behavior with sex-dependent changes in Purkinje cell firing, we reanalyzed the myoclonus data we published previously [15]. The female Sgce KO mice showed four times more myoclonus than male Sgce KO mice. These results suggest that the abnormal Purkinje cell firing in the Sgce KO mice contributes to myoclonus phenotype.
Our results expand the existing research on Purkinje cell firing in DYT11 dystonia from the shRNA-mediated knockdown [37]. The Sgce knockdown mice show a reduced firing rate similar to Sgce KO female mice and an increased CV opposite to the Sgce KO mice. It should be noted that the reported Sgce knockdown mice data are from in vivo recordings, and we used brain slice recording with both GABAergic and glutamatergic transmission blocked. Furthermore, shRNA knockdown is introduced in adult mice, while the Sgce gene was inactivated constitutively throughout the animals’ lifespan in Sgce KO mice. These differences could contribute to the discrepancy. Although the knockdown experiments used both males and females, the effect of sex on the Purkinje cell firing property was not investigated. Furthermore, we compared tonic and non-tonic cells in the current study, which is lacking in the study of Sgce knockdown mice. We extended their results by showing that female non-tonic Purkinje cells showed decreased firing frequency, likely due to decreased intrinsic excitability, and male non-tonic Purkinje cells showed a reduced CV.
Our results showed sex-specific alternations in Purkinje cell firing in the Sgce KO mice. Female Sgce KO mice showed greater changes than male Sgce KO mice. Interestingly, reanalysis of the myoclonus data showed a similar differential change in the myoclonus data. Female Sgce KO mice showed 4 times more myoclonus than male Sgce KO mice. This is consistent with the finding that the average age of onset for female DYT11 patients is 5 years versus 8 years for male patients [8] and other dystonia patient databases in general [7].
Past studies have shown that the cerebellum is critically involved in DYT11 dystonia pathogenesis [19–23]. This is further supported by brain imaging studies in DYT11 patients [25–32]. DYT11 patients show abnormal responses to cerebellar eye-blink classic conditioning [33, 34] and perform abnormally in a cerebellar saccadic adaptation task [35]. shRNA knockdown of Sgce mRNA in the cerebellum leads to motor deficits, spinning, and myoclonic-like jerky movements [37]. Our results here further support a direct role of cerebellum involvement. However, Purkinje cell-specific Sgce KO mice have no myoclonus phenotype and only exhibit motor learning deficits [10]. This suggests that myoclonus may not originate from the cerebellum but downstream of the brain network, which leads to the pathogenesis of DYT11 dystonia.
What might be the upstream brain network abnormality that drives Purkinje cell abnormality and myoclonus in DYT11 dystonia? The basal ganglia and the cerebellum are interconnected at the subcortical level with disynaptic pathways. The subthalamic nucleus in the basal ganglia connects to the cerebellar cortex via pontine nuclei [46]. We previously conditionally knocked out Sgce in the striatal medium spiny neurons using RGS9-cre mice [47, 48], and the mutant mice failed to exhibit the myoclonus phenotype [9], suggesting striatum may not be the origin. Future studies should analyze the conditional Sgce KO mice restricted to the cerebral cortex, dopaminergic neurons, and striatal cholinergic neurons, to determine the origin of myoclonus generation in DYT11 dystonia.
There are limitations associated with the current study. We used glutamatergic and GABAergic antagonists to block synaptic transmission in brain slice recording. Synaptic inputs to Purkinje cells were not measured and compared. Furthermore, we did not investigate whether altered Purkinje cell firing leads to any physiological changes elsewhere. Finally, although profound Purkinje cell firing changes in female Sgce KO mice correlate with their more pronounced myoclonus phenotype, correlation does not mean causation. Further direct experimental manipulations are needed to demonstrate the relationship.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by the Institutional Animal Care and Use Committees of the University of Florida. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
HX, PG, and FY conducted the experiments, and HX and YL wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Research reported in this publication was provided by Tyler’s Hope for a Dystonia Cure and the Norman Fixel Institute for Neurological Diseases at UF Health, National Institutes of Health grants (NS129873 and AG087418). HX, FY, and YL were partially supported by the Office of the Assistant Secretary of Defense for Health Affairs through the Peer-Reviewed Medical Research Program Discovery Award (W81XWH1810099 and W81XWH2110198).
Acknowledgments
We thank the animal care staff and undergraduate students for their technical assistance, and reviewers for their excellent suggestions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Author disclaimer
The content is solely the authors’ responsibility and does not necessarily represent the official views of the National Institutes of Health. Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Department of Defense.
Abbreviations
CV: coefficient of variation; KO: knockout; M-D: Myoclonus-dystonia; PCR: polymerase chain reaction; RMP: resting membrane potential; SEM: standard error of the mean; SGCE: human gene codes for ε-sarcoglycan; Sgce: mouse gene codes for ε-sarcoglycan; shRNA: small hairpin RNA; WT: wild-type.
References
1. Zimprich, A, Grabowski, M, Asmus, F, Naumann, M, Berg, D, Bertram, M, et al. Mutations in the gene encoding epsilon-sarcoglycan cause myoclonus-dystonia syndrome. Nat Genet (2001) 29:66–9. doi:10.1038/ng709
2. Kinugawa, K, Vidailhet, M, Clot, F, Apartis, E, Grabli, D, and Roze, E. Myoclonus-dystonia: an update. Mov Disord (2009) 24:479–89. doi:10.1002/mds.22425
3. Cazurro-Gutiérrez, A, Marcé-Grau, A, Correa-Vela, M, Salazar, A, Vanegas, MI, Macaya, A, et al. ε-Sarcoglycan: unraveling the myoclonus-dystonia gene. Mol Neurobiol (2021) 58:3938–52. doi:10.1007/s12035-021-02391-0
4. Misbahuddin, A, Placzek, M, Lennox, G, Taanman, JW, and Warner, TT. Myoclonus-dystonia syndrome with severe depression is caused by an exon-skipping mutation in the epsilon-sarcoglycan gene. Mov Disord (2007) 22:1173–5. doi:10.1002/mds.21297
5. Doheny, DO, Brin, MF, Morrison, CE, Smith, CJ, Walker, RH, Abbasi, S, et al. Phenotypic features of myoclonus-dystonia in three kindreds. Neurology (2002) 59:1187–96. doi:10.1212/wnl.59.8.1187
6. Fearon, C, Peall, KJ, Vidailhet, M, and Fasano, A. Medical management of myoclonus-dystonia and implications for underlying pathophysiology. Parkinsonism Relat Disord (2020) 77:48–56. doi:10.1016/j.parkreldis.2020.06.016
7. Kilic-Berkmen, G, Scorr, LM, McKay, L, Thayani, M, Donsante, Y, Perlmutter, JS, et al. Sex differences in dystonia. Mov Disord Clin Pract (2024) 11:973–82. doi:10.1002/mdc3.14059
8. Raymond, D, Saunders-Pullman, R, de Carvalho Aguiar, P, Schule, B, Kock, N, Friedman, J, et al. Phenotypic spectrum and sex effects in eleven myoclonus-dystonia families with epsilon-sarcoglycan mutations. Mov Disord (2008) 23:588–92. doi:10.1002/mds.21785
9. Yokoi, F, Dang, MT, Zhou, T, and Li, Y. Abnormal nuclear envelopes in the striatum and motor deficits in DYT11 myoclonus-dystonia mouse models. Hum Mol Genet (2012) 21:916–25. doi:10.1093/hmg/ddr528
10. Yokoi, F, Dang, MT, Yang, G, Li, J, Doroodchi, A, Zhou, T, et al. Abnormal nuclear envelope in the cerebellar Purkinje cells and impaired motor learning in DYT11 myoclonus-dystonia mouse models. Behav Brain Res (2012) 227:12–20. doi:10.1016/j.bbr.2011.10.024
11. Yokoi, F, Dang, MT, Mitsui, S, and Li, Y. Exclusive paternal expression and novel alternatively spliced variants of epsilon-sarcoglycan mRNA in mouse brain. FEBS Lett (2005) 579:4822–8. doi:10.1016/j.febslet.2005.07.065
12. Li, J, Liu, Y, Li, Q, Huang, X, Zhou, D, Xu, H, et al. Mutation in ε-sarcoglycan induces a myoclonus-dystonia syndrome-like movement disorder in mice. Neurosci Bull (2021) 37:311–22. doi:10.1007/s12264-020-00612-5
13. Xiao, J, Vemula, SR, Xue, Y, Khan, MM, Carlisle, FA, Waite, AJ, et al. Role of major and brain-specific Sgce isoforms in the pathogenesis of myoclonus-dystonia syndrome. Neurobiol Dis (2017) 98:52–65. doi:10.1016/j.nbd.2016.11.003
14. Maltese, M, Martella, G, Imbriani, P, Schuermans, J, Billion, K, Sciamanna, G, et al. Abnormal striatal plasticity in a DYT11/SGCE myoclonus dystonia mouse model is reversed by adenosine A2A receptor inhibition. Neurobiol Dis (2017) 108:128–39. doi:10.1016/j.nbd.2017.08.007
15. Yokoi, F, Dang, MT, Li, J, and Li, Y. Myoclonus, motor deficits, alterations in emotional responses and monoamine metabolism in epsilon-sarcoglycan deficient mice. J Biochem (2006) 140:141–6. doi:10.1093/jb/mvj138
16. Piras, G, El Kharroubi, A, Kozlov, S, Escalante-Alcalde, D, Hernandez, L, Copeland, NG, et al. Zac1 (Lot1), a potential tumor suppressor gene, and the gene for epsilon-sarcoglycan are maternally imprinted genes: identification by a subtractive screen of novel uniparental fibroblast lines. Mol Cell Biol (2000) 20:3308–15. doi:10.1128/mcb.20.9.3308-3315.2000
17. Muller, B, Hedrich, K, Kock, N, Dragasevic, N, Svetel, M, Garrels, J, et al. Evidence that paternal expression of the epsilon-sarcoglycan gene accounts for reduced penetrance in myoclonus-dystonia. Am J Hum Genet (2002) 71:1303–11. doi:10.1086/344531
18. Zhang, L, Yokoi, F, Parsons, DS, Standaert, DG, and Li, Y. Alteration of striatal dopaminergic neurotransmission in a mouse model of DYT11 myoclonus-dystonia. Plos One (2012) 7:e33669. doi:10.1371/journal.pone.0033669
19. Menozzi, E, Balint, B, Latorre, A, Valente, EM, Rothwell, JC, and Bhatia, KP. Twenty years on: myoclonus-dystonia and ε-sarcoglycan - neurodevelopment, channel, and signaling dysfunction. Mov Disord (2019) 34:1588–601. doi:10.1002/mds.27822
20. Roze, E, Lang, AE, and Vidailhet, M. Myoclonus-dystonia: classification, phenomenology, pathogenesis, and treatment. Curr Opin Neurol (2018) 31:484–90. doi:10.1097/WCO.0000000000000577
21. Ritz, K, Groen, JL, Kruisdijk, JJ, Baas, F, Koelman, JH, and Tijssen, MA. Screening for dystonia genes DYT1, 11 and 16 in patients with writer's cramp. Mov Disord (2009) 24:1390–2. doi:10.1002/mds.22632
22. Lin, WS. Translating genetic Discovery into a mechanistic understanding of pediatric movement disorders: lessons from genetic dystonias and related disorders. Adv Genet (Hoboken) (2023) 4:2200018. doi:10.1002/ggn2.202200018
23. El Atiallah, I, Bonsi, P, Tassone, A, Martella, G, Biella, G, Castagno, AN, et al. Synaptic dysfunction in dystonia: update from experimental models. Curr Neuropharmacol (2023) 21:2310–22. doi:10.2174/1570159X21666230718100156
24. Jackson, NN, Stagray, JA, and Snell, HD. Cerebellar contributions to dystonia: unraveling the role of Purkinje cells and cerebellar nuclei. Dystonia (2025) 4:14006. doi:10.3389/dyst.2025.14006
25. Nitschke, MF, Erdmann, C, Trillenberg, P, Sprenger, A, Kock, N, Sperner, J, et al. Functional MRI reveals activation of a subcortical network in a 5-year-old girl with genetically confirmed myoclonus-dystonia. Neuropediatrics (2006) 37:79–82. doi:10.1055/s-2006-924109
26. Beukers, RJ, Foncke, EM, van der Meer, JN, Nederveen, AJ, de Ruiter, MB, Bour, LJ, et al. Disorganized sensorimotor integration in mutation-positive myoclonus-dystonia: a functional magnetic resonance imaging study. Arch Neurol (2010) 67:469–74. doi:10.1001/archneurol.2010.54
27. Carbon, M, Raymond, D, Ozelius, L, Saunders-Pullman, R, Frucht, S, Dhawan, V, et al. Metabolic changes in DYT11 myoclonus-dystonia. Neurology (2013) 80:385–91. doi:10.1212/WNL.0b013e31827f0798
28. van der Meer, JN, Beukers, RJ, van der Salm, SM, Caan, MW, Tijssen, MA, and Nederveen, AJ. White matter abnormalities in gene-positive myoclonus-dystonia. Mov Disord (2012) 27:1666–72. doi:10.1002/mds.25128
29. Tarrano, C, Galléa, C, Delorme, C, McGovern, EM, Atkinson-Clement, C, Barnham, IJ, et al. Association of abnormal explicit sense of agency with cerebellar impairment in myoclonus-dystonia. Brain Commun (2024) 6:fcae105. doi:10.1093/braincomms/fcae105
30. Tarrano, C, Zito, G, Galléa, C, Delorme, C, McGovern, EM, Atkinson-Clement, C, et al. Microstructure of the cerebellum and its afferent pathways underpins dystonia in myoclonus dystonia. Eur J Neurol (2024) 31:e16460. doi:10.1111/ene.16460
31. Tarrano, C, Galléa, C, Delorme, C, McGovern, EM, Atkinson-Clement, C, Brochard, V, et al. Psychiatric phenotype in neurodevelopmental myoclonus-dystonia is underpinned by abnormality of cerebellar modulation on the cerebral cortex. Sci Rep (2024) 14:22341. doi:10.1038/s41598-024-73386-9
32. van der Salm, SM, van der Meer, JN, Nederveen, AJ, Veltman, DJ, van Rootselaar, AF, and Tijssen, MA. Functional MRI study of response inhibition in myoclonus dystonia. Exp Neurol (2013) 247:623–9. doi:10.1016/j.expneurol.2013.02.017
33. Popa, T, Milani, P, Richard, A, Hubsch, C, Brochard, V, Tranchant, C, et al. The neurophysiological features of myoclonus-dystonia and differentiation from other dystonias. JAMA Neurol (2014) 71:612–9. doi:10.1001/jamaneurol.2014.99
34. Weissbach, A, Werner, E, Bally, JF, Tunc, S, Löns, S, Timmann, D, et al. Alcohol improves cerebellar learning deficit in myoclonus-dystonia: a clinical and electrophysiological investigation. Ann Neurol (2017) 82:543–53. doi:10.1002/ana.25035
35. Hubsch, C, Vidailhet, M, Rivaud-Péchoux, S, Pouget, P, Brochard, V, Degos, B, et al. Impaired saccadic adaptation in DYT11 dystonia. J Neurol Neurosurg Psychiatry (2011) 82:1103–6. doi:10.1136/jnnp.2010.232793
36. Sadnicka, A, Galea, JM, Chen, JC, Warner, TT, Bhatia, KP, Rothwell, JC, et al. Delineating cerebellar mechanisms in DYT11 myoclonus-dystonia. Mov Disord (2018) 33:1956–61. doi:10.1002/mds.27517
37. Washburn, S, Fremont, R, Moreno-Escobar, MC, Angueyra, C, and Khodakhah, K. Acute cerebellar knockdown of Sgce reproduces salient features of myoclonus-dystonia (DYT11) in mice. Elife (2019) 8:e52101. doi:10.7554/eLife.52101
38. Voelkl, B, Altman, NS, Forsman, A, Forstmeier, W, Gurevitch, J, Jaric, I, et al. Reproducibility of animal research in light of biological variation. Nat Rev Neurosci (2020) 21:384–93. doi:10.1038/s41583-020-0313-3
39. Liu, Y, Xing, H, Ernst, AF, Liu, C, Maugee, C, Yokoi, F, et al. Hyperactivity of Purkinje cell and motor deficits in C9orf72 knockout mice. Mol Cell Neurosci (2022) 121:103756. doi:10.1016/j.mcn.2022.103756
40. Lyu, S, Xing, H, DeAndrade, MP, Perez, PD, Yokoi, F, Febo, M, et al. The role of BTBD9 in the cerebellum, sleep-like behaviors and the restless legs syndrome. Neuroscience (2020) 440:85–96. doi:10.1016/j.neuroscience.2020.05.021
41. Lyu, S, Xing, H, Liu, Y, Girdhar, P, Yokoi, F, and Li, Y. Further studies on the role of BTBD9 in the cerebellum, sleep-like behaviors and the restless legs syndrome. Neuroscience (2022) 505:78–90. doi:10.1016/j.neuroscience.2022.10.008
42. Xing, H, Girdhar, P, Liu, Y, Yokoi, F, Vaillancourt, DE, and Li, Y. Subtle changes in Purkinje cell firing in Purkinje cell-specific Dyt1 ΔGAG knock-in mice. Dystonia (2025) 4. doi:10.3389/dyst.2025.14148
43. Liu, Y, Xing, H, Wilkes, BJ, Yokoi, F, Chen, H, Vaillancourt, DE, et al. The abnormal firing of Purkinje cells in the knockin mouse model of DYT1 dystonia. Brain Res Bull (2020) 165:14–22. doi:10.1016/j.brainresbull.2020.09.011
44. Womack, MD, and Khodakhah, K. Characterization of large conductance Ca2+-activated K+ channels in cerebellar Purkinje neurons. Eur J Neurosci (2002) 16:1214–22. doi:10.1046/j.1460-9568.2002.02171.x
45. Tian, J, Tep, C, Benedick, A, Saidi, N, Ryu, JC, Kim, ML, et al. p75 regulates Purkinje cell firing by modulating SK channel activity through Rac1. J Biol Chem (2014) 289:31458–72. doi:10.1074/jbc.M114.589937
46. Bostan, AC, and Strick, PL. The basal ganglia and the cerebellum: nodes in an integrated network. Nat Rev Neurosci (2018) 19:338–50. doi:10.1038/s41583-018-0002-7
47. Dang, MT, Yokoi, F, Yin, HH, Lovinger, DM, Wang, Y, and Li, Y. Disrupted motor learning and long-term synaptic plasticity in mice lacking NMDAR1 in the striatum. Proc Natl Acad Sci USA (2006) 103:15254–9. doi:10.1073/pnas.0601758103
Keywords: Purkinje cells, dystonia, SGCE, DYT11, electrophysiology
Citation: Xing H, Girdhar P, Yokoi F and Li Y (2025) Sex-specific alterations of Purkinje cell firing in Sgce knockout mice and correlations with myoclonus. Dystonia 4:14415. doi: 10.3389/dyst.2025.14415
Received: 29 January 2025; Accepted: 06 March 2025;
Published: 18 March 2025.
Edited by:
Roy Sillitoe, Baylor College of Medicine, United StatesCopyright © 2025 Xing, Girdhar, Yokoi and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuqing Li, eXVxaW5nbGlAdWZsLmVkdQ==