 Angela Wong
Angela Wong Qiuyu Sun1
Qiuyu Sun1 Qutuba G. Karwi
Qutuba G. Karwi- 1Department of Pediatrics, Faculty of Medicine and Dentistry, University of Alberta, Edmonton, AB, Canada
- 2Department of Microbiology, College of Medicine, University of Diyala, Baqubaa, Diyala, Iraq
- 3Division of BioMedical Sciences, Faculty of Medicine, Memorial University of Newfoundland, Saint John’s, NL, Canada
Recent literature extensively investigates the crucial role of energy metabolism in determining the inflammatory response and polarization status of macrophages. This rapidly expanding area of research highlights the importance of understanding the link between energy metabolism and macrophage function. The metabolic pathways in macrophages are intricate and interdependent, and they can affect the polarization of macrophages. Previous studies suggested that glucose flux through cytosolic glycolysis is necessary to trigger pro-inflammatory phenotypes of macrophages, and fatty acid oxidation is crucial to support anti-inflammatory responses. However, recent studies demonstrated that this understanding is oversimplified and that the metabolic control of macrophage polarization is highly complex and not fully understood yet. How the metabolic flux through different metabolic pathways (glycolysis, glucose oxidation, fatty acid oxidation, ketone oxidation, and amino acid oxidation) is altered by obesity- and type 2 diabetes (T2D)-associated insulin resistance is also not fully defined. This mini-review focuses on the impact of insulin resistance in obesity and T2D on the metabolic flux through the main metabolic pathways in macrophages, which might be linked to changes in their inflammatory responses. We closely evaluated the experimental studies and methodologies used in the published research and highlighted priority research areas for future investigations.
Introduction
Obesity and type 2 diabetes (T2D) are conditions marked by insulin resistance and persistent low-grade inflammation [1–3]. Chronic tissue inflammation is now recognized as an essential characteristic of obesity and T2D, affecting insulin-target tissues such as adipose tissue, liver, muscle, and heart. The recruitment, accumulation, and activation of pro-inflammatory macrophages in metabolic tissues play important roles in driving this chronic low-grade inflammation. Although other types of immune cells also contribute to these inflammatory processes, macrophages are primary effector cells known to be closely associated with the development of cardiometabolic disease, including obesity and T2D (see for [3–7] review). Macrophages are crucial immune cells involved in immune response [8] and play important roles in tissue repair and maintaining the body’s homeostasis [9]. The inflammatory responses of macrophages are supported by different metabolic pathways (glycolysis, glucose oxidation, fatty acid oxidation, ketone oxidation, and amino acid oxidation) that adjust to their polarization state [10]. Importantly, the metabolic flux through different metabolic pathways could be influenced by the tissue microenvironment, the availability of oxidative substrates, and neurohormonal status. In addition, altered macrophage energy metabolism can impact tissue repair, inflammatory responses, and the severity of insulin resistance [2, 11–13].
Earlier studies suggested that pro-inflammatory (also called M1-like or classical) macrophages are highly glycolytic, while anti-inflammatory (also called M2-like or alternative) macrophages are highly oxidative [14–16]. However, this classification has been challenged as simplistic, as emerging evidence shows that macrophages have a very complex and dynamic metabolic profile that influences their activity. Importantly, “immunometabolism” has emerged as a prerequisite trigger of macrophage activation and phenotype. Nevertheless, minimal research has been steered to understand how the metabolic phenotype of macrophages influences disease progression [17]. Therefore, a better understanding of macrophage metabolism may shed an innovative light on the pathological basis of disease and lead to the future development of macrophage-targeted treatment approaches. In this mini-review, we discussed how carbon flux through the main metabolic pathways in macrophages is perturbed in obesity and T2D and how that influences the inflammatory response, activity and metabolic profile of macrophages.
Glycolysis
Insulin signalling is a key modulator of macrophage metabolism by regulating glucose uptake and oxidation [18, 19]. However, in obesity and T2D, the insulin signalling pathway is impaired, leading to insulin resistance in macrophages [18, 19]. Interestingly, this results in the upregulation of glycolysis and glucose uptake, which is associated with proinflammatory macrophage polarization [20]. This metabolic switch is crucial for the host defence mechanisms of macrophages, such as cytokine production and phagocytosis. Increased glucose influx in insulin-resistant macrophages is facilitated by upregulated glucose transporter 1 (GLUT1) expression [21, 22]. In high-fat diet (HFD) fed mice, the overexpression of GLUT1 in the pro-inflammatory macrophages also led to a hyperinflammatory state with the elevated secretion of inflammatory mediators and increased reactive oxygen species (ROS) production [23]. The metabolomic analysis also demonstrated an increased glucose uptake in the GLUT1 overexpressed macrophages enhances glucose flux through the pentose phosphate pathway, where glucose is utilized to generate NADPH for use in biosynthetic pathways and ROS production [23]. Upon activation, proinflammatory macrophages undergo a “respiratory burst,” also called “glycolytic burst,” driven by augmented NADPH oxidase activity to generate large amounts of ROS as a defence mechanism against pathogens [24]. To support redox balance, the glycolysis-PPP axis is triggered in response to M1 polarization, presumably to support the increased generation of NADPH for use by NADPH oxidase as well as glutathione production used by macrophages to protect from the excessive amounts of superoxide being produced [24]. This suggests that GLUT1-mediated glucose metabolism plays an important part in driving the pro-inflammatory state of macrophages in obesity and T2D (Figure 1). Consistent with that, genetic deletion of GLUT1 in bone marrow-derived macrophages (BMDMs) displays marked reductions in the classically activated pro-inflammatory markers and associated oxidative stress [25]. Inhibiting glycolysis or treating macrophages with an antioxidant (N-acetyl-cysteine) reversed GLUT1-mediated pro-inflammatory elevations [23].
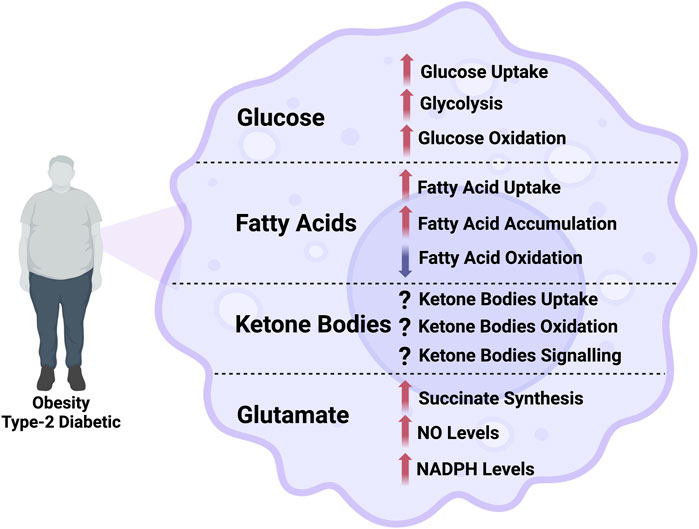
Figure 1. Illustration of the alterations in the flux through different metabolic pathways in macrophage in obesity and type-2 diabetes. NO, nitric oxide; NADPH, nicotinamide adenine dinucleotide phosphate.
Increased glucose availability in T2D-induced hyperglycemic conditions can also promote the formation of advanced glycation end products (AGEs) in macrophages [26, 27]. AGEs are pathogenic factors that trigger the activation of a number of signalling pathways in macrophages, including the NF- κB and the MAPK signalling pathways under hyperglycemia conditions [28, 29]. Enhanced AGEs in BMDMs increase interleukin 6 (IL-6) and tumour necrosis factor-alpha (TNF-α) production [30]. AGEs also enhance the polarization of macrophages toward the pro-inflammatory state by inducing the expression of pro-inflammatory molecules in T2D [29, 31]. Hypoxia-inducible factor 1 subunit alpha (HIF1α) also plays a critical role in increasing glycolytic flux and abrogating oxidative metabolism (OXPHOS) in macrophages in obesity and T2D [32, 33]. To further support this, HIF-1α gene deletion in mice protected against HFD-induced adipose tissue inflammation and systemic insulin resistance [34].
Glucose oxidation
Increasing glucose uptake in macrophages by overexpressing GLUT1 enhances glucose flux through cytosolic glycolysis and mitochondrial glucose oxidation [23]. While these metabolic changes are associated with promoting the pro-inflammatory phenotype of macrophages, it is still unclear whether the increase in glycolysis and/or glucose oxidation are essential for promoting pro-inflammatory macrophages. Min et al. demonstrated that pyruvate dehydrogenase kinase (PDK), which inhibits the pyruvate dehydrogenase-mediated conversion of cytosolic pyruvate to mitochondrial acetyl-CoA, functions as a metabolic checkpoint in inflammatory macrophages [35]. Deletion of PDK2 and PDK4 completely abolishes the development of pro-inflammatory macrophages in HFD-induced insulin resistance [35]. Inhibition of macrophage glucose oxidation is also associated with weight loss, reduced insulin resistance, and decreased adipose tissue inflammation [35]. Taken together, inhibiting macrophage glucose oxidation is a potential target to limit the severity of insulin resistance in obesity and T2D (Figure 1).
It has been suggested that the neurogenic locus notch homolog protein 1 (NOTCH1) signalling pathway contributes to the activation of mitochondrial glucose oxidation in obesity. For instance, Xu et al. showed an enhanced macrophage glucose oxidation in obesity that is mediated, at least in part, by increased recruitment of the NOTCH1 intracellular domain (NICD1) to nuclear and mitochondrial genes that encode respiratory chain components [36]. This effect also involved NOTCH-dependent induction of pyruvate dehydrogenase phosphatase 1 (Pdp1) expression, pyruvate dehydrogenase activity, and glucose flux to the tricarboxylic acid (TCA) cycle [36]. This enhancement of glucose oxidation is associated with augmented levels of mitochondrial DNA transcription in pro-inflammatory macrophages, thus causing enhanced mitochondrial ROS levels [36]. Therefore, glucose oxidation may be a target in macrophages to alleviate insulin resistance and inflammation induced by HFD.
Fatty acid oxidation
Obesity and T2D-induced lipid accumulation in adipose tissue are associated with elevated fatty acid uptake, increased macrophage infiltration, and decreased fatty acid oxidation [37, 38]. Moreover, increased lipolysis at the adipose tissue level has been linked to lipid-droplet accumulation in adipose tissue macrophages (ATMs) and obesity-induced inflammation [39, 40]. Studies have shown that macrophages develop a distinct phenotype in obesity exemplified by increased lysosomal acid type lipase, fatty acid receptor, ATP-binding cassette A1 expressions, and inflammatory cytokines (IL-1β and TNF-α) [41, 42]. In addition to acting as fuel for activated macrophages, excessive lipid intake is also shown to be a primary factor that causes pro-inflammatory macrophage polarization in obesity and T2D [43].
The fatty acid translocase CD36 binds to fatty acids and is important for fatty acid uptake at the myocardium, skeletal muscle, gastrointestinal tract, liver, and adipose tissue level [44–46]. In macrophages, CD36 primarily acts as a scavenger receptor, recognizing specific self and nonself molecular patterns and triggering internalization and inflammatory signalling pathways to eliminate pathogens and altered self components, such as apoptotic cells [47]. CD36 cooperates with toll-like receptor (TLR)-4 and −6 to trigger inflammatory responses to altered self-components oxidized LDL (ox-LDL) and amyloid-β [48]. CD36 also acts as a coreceptor with TLR2 and −6 in recognizing microbial diacylglycerides [49]. The deletion of CD36 in BMDMs displayed improved insulin signalling and reduced macrophage infiltration in adipose tissue [50, 51]. This may be attributed to the potential role of upregulated fatty acid uptake in mediating obesity-induced inflammation and insulin resistance (Figure 1), although this is yet to be directly investigated. Recent studies have shown that CD36 is important in the mitochondrial metabolic switch from oxidative phosphorylation to superoxide production in response to ox-LDL and that mitochondrial reactive oxygen species positively correlate with macrophage CD36 expression [52].
Fatty acid oxidation occurs within the mitochondria, and several steps are required to activate the fatty acids and transport them into the mitochondria [53–55]. Cytosolic fatty acids are first esterified to fatty acyl-CoA (a process consuming two high-energy phosphate bonds as ATP is converted to AMP), followed by the transfer of the fatty acid moiety to carnitine via the action of carnitine palmitoyl-transferase 1 (CPT1) to form fatty acylcarnitine. CPT1, residing on the outer mitochondrial membrane, works collaboratively with carnitine acyltranslocase and carnitine palmitoyl-transferase 2 (CPT2), residing on the inner mitochondrial member, to transfer fatty acylcarnitine into the mitochondrial matrix, where it is converted back to fatty acyl-CoA. These acyl-CoAs then undergo β-oxidation to produce reduced equivalents (NADH and FADH2) for the electron transport chain (ETC), as well as acetyl-CoA for the tricarboxylic acid (TCA) cycle. Malonyl-CoA can be generated by acetyl-CoA carboxylase (ACC) from cytosolic acetyl-CoA [56, 57]. Malonyl-CoA can be converted back to acetyl-CoA by malonyl-CoA decarboxylase (MCD) [58, 59]. While the role of ACC enzymes (ACC1 and ACC2) in macrophages is not fully defined, individual deletion of ACC1 or ACC2 in the myeloid lineage is a prerequisite for the function of highly proliferative T cells, but not for macrophages [60]. Recent studies have shown that ACC is required for the early metabolic switch to glycolysis and remodelling of the fatty acid metabolism in macrophages. Using mice with myeloid-specific deletion of both ACC isoforms, ACC deficiency impairs macrophage innate immune functions, including bacterial clearance [61]. Myeloid-specific deletion or pharmacological inhibition of ACC in mice attenuated LPS-induced expression of proinflammatory cytokines interleukin-6 (IL-6) and IL-1β. In contrast, pharmacological inhibition of ACC increased susceptibility to bacterial peritonitis in wild-type mice [61].
It has been shown that the overabundant influx of fatty acids largely shifts from fatty acid oxidation to triglyceride, phospholipid, and ceramide synthesis, contributing to macrophage lipotoxicity [62–64]. This also contributes to macrophage insulin resistance and the consequential promotion of mitochondrial dysfunction in macrophages [63]. While indirect evidence suggests that altered fatty acid metabolism influences macrophage activation in obesity and T2D [65], it is unknown whether macrophage fatty acid oxidation is upregulated or downregulated in obesity and T2D. Malandrino et al. reported that enhancing fatty acid oxidation in human ATMs reduces ROS, endoplasmic reticulum stress, and pro-inflammatory responses of macrophages [37]. In line with that, the deletion of macrophage carnitine palmitoyl transferase 1A (CPT1A) catalyzes the transfer of the long-chain acyl group in acyl-CoA ester to carnitine. This allows fatty acids to enter the mitochondrial matrix for oxidation and exacerbates the accumulation of diacylglycerols and triacylglycerols after palmitate treatment in vitro [66]. CPT1A deletion also increased pro-inflammatory signalling, cytokine expression and endoplasmic reticulum stress after palmitate treatment [66]. Consistent with that, decreasing triglyceride and free cholesterol levels in macrophages mitigates the activation of pro-inflammatory macrophages, supporting the link between lipid accumulation in these cells and the switch to the pro-inflammatory polarization state (Figure 1) [43]. The intermediates of the biosynthetic pathways for triacylglycerol or phospholipids can affect the inflammatory and insulin signalling pathways in different tissues. Saturated fatty acids are also precursors of sphingolipids; in particular, ceramides are strongly linked to insulin resistance and inflammation. TLR4 signalling can trigger ceramide biosynthesis, promoting insulin resistance by activating protein phosphatase 2A and protein kinase C-zeta, ultimately inhibiting Akt [67]. Ceramides may also activate the inflammasome, inducing IL-1β secretion in macrophages, which can blunt insulin signalling [68]. These results might suggest that enhancing macrophage fatty acid oxidation could reduce macrophage activation and mitigate insulin resistance in obesity and T2D. However, this needs to be directly investigated in future research.
Insulin-resistant adipocytes also release greater levels of fatty acids while activating ATMs, leading to an intensified cycle of inflammation through the indirect stimulation of the macrophage toll-like receptor 4 (TLR4) [69, 70]. This causes the initiation of the Jun N-terminal kinase and inhibitor of κB kinase (JNK/IKK- κB) pathways followed by inflammatory cascades [71]. Suganami et al. showed that coculturing obese mice-derived hypertrophied adipocytes and macrophages augments the production of TNF-α in macrophages [69]. The released TNF- α in turn promotes the secretion of FFAs and inflammatory changes in adipocytes [69]. To further support this, TLR4-deletion in BMDMs inhibits saturated FA-induced inflammation via inhibiting palmitate-induced activation of the JNK signalling pathway [72]. Therefore, this shift in fatty acid metabolism towards greater production of inflammatory lipids and levels of FFAs in ATMs exacerbate insulin resistance in obesity and T2D [73, 74]. Glucose-6-phosphate dehydrogenase (G6PD) is a key enzyme that produces cellular NADPH, which is required for cellular redox potential and the biosynthesis of fatty acids and cholesterol. Macrophage G6PD levels are increased in the adipose tissue of obese animals, and G6PD mRNA levels positively correlated with those of pro-inflammatory genes [75]. Lipopolysaccharide (LPS) and free fatty acids, which initiate pro-inflammatory signals, stimulated macrophage G6PD. Overexpression of macrophage G6PD potentiated the expression of pro-inflammatory and pro-oxidative genes responsible for the aggravation of insulin sensitivity in adipocytes. Macrophage G6PD stimulates the p38 mitogen-activated protein kinase (MAPK) and NF-κB pathways, causing a vicious cycle of oxidative stress and pro-inflammatory cascade [75].
Macrophage infiltration to the myocardium also increases due to obesity and T2D-induced inflammation. Saturated fatty acids contribute to myocardial inflammation by inducing the release of inflammatory molecules through pattern recognition receptors (PRRs) in macrophages [71, 72, 76]. Enhanced fatty acid delivery to the heart in obesity and T2D causes uncoupling of fatty acid oxidation from adenosine diphosphate phosphorylation and promotes further mitochondrial dysfunction in cardiomyocytes [77–80]. The imbalance in cardiac lipid metabolism leads to the accumulation of ceramides and diacylglycerols in the hearts of obese and diabetic patients [77, 81–83]. Ceramides activate the NLRP3 inflammasome and further promote cardiac lipotoxicity in palmitate-exposed human cardiac cells and HFD-fed mice [84]. The interplay between altered cardiac fatty acid oxidation and macrophage infiltration into the myocardium in obesity and T2D is an interesting scope for future investigations.
Ketone oxidation
Ketone bodies are organic compounds mainly produced by the liver by breaking down fatty acid molecules. The three major ketone bodies are β-hydroxybutyrate (βOHB), acetoacetate (AcAc), and acetone. βOHB is first oxidized to AcAc by βOHB dehydrogenase, followed by conversion to acetoacetyl-CoA by succinyl-CoA:3 oxoacid-CoA transferase (SCOT). The end-product of ketone oxidation is acetyl-CoA, which has a similar fate as acetyl-CoA produced from fatty acid or glucose oxidation. While there is limited data regarding how ketone metabolism is regulated in macrophages and how it might be altered in obesity and T2D, a recent study using isotope tracking LC/MS untargeted metabolomics showed that macrophages could oxidize ketones with preferential utilization of AcAc compared to βOHB [85]. Preclinical studies have shown that enhancing circulating AcAc levels ameliorates diet-induced hepatic fibrosis, and this protective effect is abolished in macrophage-specific SCOT knock-out mice [85]. These findings suggest that increasing macrophage ketone oxidation plays a critical role in modifying the inflammatory responses of macrophages [85].
Furthermore, another study demonstrated that administration of βOHB increases the expression of IL-10 and arginase 1, markers of the inflammation-resolving state of macrophages and the resolution of damaged intestinal tissue in a mouse model of inflammatory bowel disease [86]. In addition to supporting macrophage energetics, βOHB could also influence macrophage activity by acting as a signalling molecule (see for [87] review). For instance, the inhibitory effect of βOHB on the NLRP3 inflammasome in BMDMs is mediating by acting as a ligand of macrophage GPR109A, a member of the hydrocarboxylic acid GPR sub-family expressed in adipose tissues (white and brown) and immune cells [88]. While these findings suggest that ketone bodies elicit a predominantly anti-inflammatory response, augmented circulating ketone levels in diabetic patients may trigger a pro-inflammatory response (Figure 1) [89–91]. Future studies are required to delineate whether this modulatory effect of ketones on macrophage function is mediated by enhancing increased macrophage ketone oxidation and/or via acting as a signalling molecule [87]. It would also be important to determine how modulating macrophage ketone oxidation affects macrophage function in obesity and T2D. Taken together, this encouraging emerging evidence suggests that ketone bodies have an inhibitory effect on the inflammatory response of macrophages, which might be beneficial against the low-grade chronic inflammation in obesity and T2D and its detrimental effects. However, it is yet to be determined whether ketone bodies modulate macrophage responses by serving as oxidative substrates to modulate macrophage ATP production or acting as signalling molecules.
Amino acid oxidation
The flux through different amino acid metabolic pathways changes according to macrophage phenotype. For instance, arginine is converted to nitric oxide (NO) via inducible NO synthase (iNOS) in pro-inflammatory M1-like macrophages [92, 93]. In contrast, it is converted to proline and polyamines via arginase-1 in inflammation-resolving M2-like macrophages (Figure 1) [94]. Glutamine is the most abundant amino acid in the body and acts as a main source of carbon and nitrogen for cells. While serum glutamine levels are lower in patients with obesity and diabetes [95], studies have demonstrated that glutamine metabolism is altered in macrophages on exposure to M1-or M2-polarizing agents. For instance, glutamine is channelled into the TCA cycle for synthesizing succinate in M1-like macrophages, leading to a marked intracellular accumulation of succinate, enhancing proinflammatory cytokine production [33]. On the contrary, glutamine is critical for acquiring the M2 polarization state. It is mostly converted to α-ketoglutarate in M2-like macrophages and enhances the production of key anti-inflammatory cytokines through its role in protein glycosylation (Figure 1) [96, 97]. Consistent with that, glutamine deprivation impairs the expression of M2-like macrophage markers in vitro [97]. Macrophages collected from obese insulin-resistant Zucker rats had a significantly lower NO production than those collected from lean control rats [98]. Interestingly, incubating macrophages from obese insulin-resistant Zucker rats with glutamine increases NO production [98]. Since NO is produced using arginine as a precursor, these findings suggest crosstalk between arginine and glutamine metabolism in macrophages, highlighting the importance of differential use of amino acids to modulate macrophage responses in insulin resistance conditions. It is important to mention that the action of iNOS in converting arginine to NO depends on the availability of an important cofactor, NADPH. Macrophages can utilize glucose and glutamine to synthesize NADPH [99]. Macrophages collected from insulin-resistant rats have impaired NO production due to impaired synthesis of NADPH and less activation of iNOS in the absence of glutamine [98]. Therefore, suggesting that glutamate contributes to NADPH synthesis in insulin-resistant macrophages seems plausible.
Augmented levels of branched-chain amino acids (BCAAs), namely leucine, isoleucine, and valine, and their respective metabolites, namely branched-chain keto acids (BCKAs), have been linked with metabolic alterations, insulin resistance, and a predisposition to T2D [100]. It has been shown that BCAAs play a role in modulating inflammatory responses in immune cells. However, the data regarding whether high levels of BCAAs promote pro-inflammatory or anti-inflammatory immune cells are inconclusive. For instance, enhancing BCAA levels promotes oxidative stress, inflammation and human peripheral blood mononuclear cell migration [101]. In contrast, high levels of BCAA exert anti-inflammatory and anti-genotoxic activity in LPS-stimulated macrophages [102]. Whether these effects are mediated via enhancing BCAAs and BCKAs contributions to mitochondrial oxidative metabolism as fuel or via acting as signalling molecules in macrophages remains to be determined in future investigations.
Discussion
Recent studies have linked macrophage metabolic processes to their inflammatory behaviour. They demonstrated that macrophages could switch from promoting tissue protection to contributing to disease development by altering the flux through different metabolic pathways. Although macrophage insulin signalling is impaired in obesity and T2D, insulin-resistant macrophages have increased glucose uptake, glycolysis, and glucose oxidation. Macrophage fatty acid uptake is also increased, but it seems uncoupled to fatty acid oxidation, which is decreased in obesity and T2D. Instead, fatty acids are converted to triacylglycerol and ceramide accumulation, which contribute to lipotoxicity in insulin-resistant macrophages. Arginine and glutamate metabolism also have divergent effects in pro- and anti-inflammatory macrophages in obesity and T2D. Understanding the complex metabolic profile of different macrophage phenotypes will help characterize how different oxidative substrates could influence macrophage responses. In addition, understanding the metabolic reprogramming behind macrophage responses will help identify new avenues for therapeutic intervention to combat obesity and T2D.
Author contributions
All authors contributed to the study’s conception and design. AW and QS performed a systematic review and critically appraised the literature. They wrote the first draft of the manuscript, and all authors commented on previous versions. All authors contributed to the article and approved the submitted version.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. QK is funded by a Janeway Foundation Research Grant and a Medical Research Fund Cox Award.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Xu, H, Barnes, GT, Yang, Q, Tan, G, Yang, D, Chou, CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest (2003) 112(12):1821–30. doi:10.1172/jci200319451
2. Olefsky, JM, and Glass, CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol (2010) 72:219–46. doi:10.1146/annurev-physiol-021909-135846
3. Chawla, A, Nguyen, KD, and Goh, YP. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol (2011) 11(11):738–49. doi:10.1038/nri3071
4. Qu, L, Matz, AJ, Karlinsey, K, Cao, Z, Vella, AT, and Zhou, B. Macrophages at the crossroad of meta-inflammation and inflammaging. Genes (Basel) (2022) 13(11):2074. doi:10.3390/genes13112074
5. Kahn, SE, Hull, RL, and Utzschneider, KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature (2006) 444(7121):840–6. doi:10.1038/nature05482
6. Czech, MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med (2017) 23(7):804–14. doi:10.1038/nm.4350
7. Wondmkun, YT. Obesity, insulin resistance, and type 2 diabetes: associations and therapeutic implications. Diabetes Metab Syndr Obes Targets Ther (2020) 13:3611–6. doi:10.2147/dmso.s275898
8. Weisberg, SP, McCann, D, Desai, M, Rosenbaum, M, Leibel, RL, and Ferrante, AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest (2003) 112(12):1796–808. doi:10.1172/jci19246
9. Rasheed, A, and Rayner, KJ. Macrophage responses to environmental stimuli during homeostasis and disease. Endocr Rev (2021) 42(4):407–35. doi:10.1210/endrev/bnab004
10. Wculek, SK, Dunphy, G, Heras-Murillo, I, Mastrangelo, A, and Sancho, D. Metabolism of tissue macrophages in homeostasis and pathology. Cell Mol Immunol (2022) 19(3):384–408. doi:10.1038/s41423-021-00791-9
11. Jung, S-B, Choi, MJ, Ryu, D, Yi, H-S, Lee, SE, Chang, JY, et al. Reduced oxidative capacity in macrophages results in systemic insulin resistance. Nat Commun (2018) 9(1):1551. doi:10.1038/s41467-018-03998-z
12. Phan, AT, Goldrath, AW, and Glass, CK. Metabolic and epigenetic coordination of T cell and macrophage immunity. Immunity (2017) 46(5):714–29. doi:10.1016/j.immuni.2017.04.016
13. Jones, RG, and Pearce, EJ. MenTORing immunity: mTOR signaling in the development and function of tissue-resident immune cells. Immunity (2017) 46(5):730–42. doi:10.1016/j.immuni.2017.04.028
14. Newsholme, P, Gordon, S, and Newsholme, EA. Rates of utilization and fates of glucose, glutamine, pyruvate, fatty acids and ketone bodies by mouse macrophages. Biochem J (1987) 242(3):631–6. doi:10.1042/bj2420631
15. Rodriguez-Prados, JC, Traves, PG, Cuenca, J, Rico, D, Aragones, J, Martin-Sanz, P, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol (2010) 185(1):605–14. doi:10.4049/jimmunol.0901698
16. Nomura, M, Liu, J, Rovira, II, Gonzalez-Hurtado, E, Lee, J, Wolfgang, MJ, et al. Fatty acid oxidation in macrophage polarization. Nat Immunol (2016) 17(3):216–7. doi:10.1038/ni.3366
17. O’Neill, LA, and Pearce, EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med (2016) 213(1):15–23. doi:10.1084/jem.20151570
18. Kubota, T, Inoue, M, Kubota, N, Takamoto, I, Mineyama, T, Iwayama, K, et al. Downregulation of macrophage Irs2 by hyperinsulinemia impairs IL-4-indeuced M2a-subtype macrophage activation in obesity. Nat Commun (2018) 9(1):4863. doi:10.1038/s41467-018-07358-9
19. Goldstein, BJ. Insulin resistance as the core defect in type 2 diabetes mellitus. Am J Cardiol (2002) 90(5):3–10. doi:10.1016/s0002-9149(02)02553-5
20. Watanabe, R, Hilhorst, M, Zhang, H, Zeisbrich, M, Berry, GJ, Wallis, BB, et al. Glucose metabolism controls disease-specific signatures of macrophage effector functions. JCI insight (2018) 3(20):e123047. doi:10.1172/jci.insight.123047
21. Lin, Y, Bai, M, Wang, S, Chen, L, Li, Z, Li, C, et al. Lactate is a key mediator that links obesity to insulin resistance via modulating cytokine production from adipose tissue. Diabetes (2022) 71(4):637–52. doi:10.2337/db21-0535
22. Lachmandas, E, Boutens, L, Ratter, JM, Hijmans, A, Hooiveld, GJ, Joosten, LA, et al. Microbial stimulation of different Toll-like receptor signalling pathways induces diverse metabolic programmes in human monocytes. Nat Microbiol (2016) 2:16246. doi:10.1038/nmicrobiol.2016.246
23. Freemerman, AJ, Johnson, AR, Sacks, GN, Milner, JJ, Kirk, EL, Troester, MA, et al. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem (2014) 289(11):7884–96. doi:10.1074/jbc.m113.522037
24. Sheppard, FR, Kelher, MR, Moore, EE, McLaughlin, NJ, Banerjee, A, and Silliman, CC. Structural organization of the neutrophil NADPH oxidase: phosphorylation and translocation during priming and activation. J Leukoc Biol (2005) 78(5):1025–42. doi:10.1189/jlb.0804442
25. Freemerman, AJ, Zhao, L, Pingili, AK, Teng, B, Cozzo, AJ, Fuller, AM, et al. Myeloid slc2a1-deficient murine model revealed macrophage activation and metabolic phenotype are fueled by GLUT1. J Immunol (2019) 202(4):1265–86. doi:10.4049/jimmunol.1800002
26. Khalid, M, Petroianu, G, and Adem, A. Advanced glycation end products and diabetes mellitus: mechanisms and perspectives. Biomolecules (2022) 12(4):542. doi:10.3390/biom12040542
27. Cai, W, Ramdas, M, Zhu, L, Chen, X, Striker, GE, and Vlassara, H. Oral advanced glycation endproducts (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin 1. Proc Natl Acad Sci U S A (2012) 109(39):15888–93. doi:10.1073/pnas.1205847109
28. Kong, X, Ma, MZ, Huang, K, Qin, L, Zhang, HM, Yang, Z, et al. Increased plasma levels of the methylglyoxal in patients with newly diagnosed type 2 diabetes 2. J Diabetes (2014) 6(6):535–40. doi:10.1111/1753-0407.12160
29. He, S, Hu, Q, Xu, X, Niu, Y, Chen, Y, Lu, Y, et al. Advanced glycation end products enhance M1 macrophage polarization by activating the MAPK pathway. Biochem Biophysical Res Commun (2020) 525(2):334–40. doi:10.1016/j.bbrc.2020.02.053
30. Jin, X, Yao, T, Zhou, Z, Zhu, J, Zhang, S, Hu, W, et al. Advanced glycation end products enhance macrophages polarization into M1 phenotype through activating RAGE/NF-κB pathway. Biomed Res Int (2015) 2015:732450. doi:10.1155/2015/732450
31. Guo, Y, Lin, C, Xu, P, Wu, S, Fu, X, Xia, W, et al. AGEs induced autophagy impairs cutaneous wound healing via stimulating macrophage polarization to M1 in diabetes. Sci Rep (2016) 6:36416. doi:10.1038/srep36416
32. Sharma, M, Boytard, L, Hadi, T, Koelwyn, G, Simon, R, Ouimet, M, et al. Enhanced glycolysis and HIF-1α activation in adipose tissue macrophages sustains local and systemic interleukin-1β production in obesity. Sci Rep (2020) 10(1):5555. doi:10.1038/s41598-020-62272-9
33. Tannahill, GM, Curtis, AM, Adamik, J, Palsson-McDermott, EM, McGettrick, AF, Goel, G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature (2013) 496(7444):238–42. doi:10.1038/nature11986
34. Takikawa, A, Mahmood, A, Nawaz, A, Kado, T, Okabe, K, Yamamoto, S, et al. HIF-1α in myeloid cells promotes adipose tissue remodeling toward insulin resistance. Diabetes (2016) 65(12):3649–59. doi:10.2337/db16-0012
35. Min, BK, Park, S, Kang, HJ, Kim, DW, Ham, HJ, Ha, CM, et al. Pyruvate dehydrogenase kinase is a metabolic checkpoint for polarization of macrophages to the M1 phenotype. Front Immunol (2019) 10:944. doi:10.3389/fimmu.2019.00944
36. Xu, J, Chi, F, Guo, T, Punj, V, Lee, WP, French, SW, et al. NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation. J Clin Invest (2015) 125(4):1579–90. doi:10.1172/jci76468
37. Malandrino, MI, Fucho, R, Weber, M, Calderon-Dominguez, M, Mir, JF, Valcarcel, L, et al. Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid-induced triglyceride accumulation and inflammation. Am J Physiology-Endocrinology Metab (2015) 308(9):E756–69. doi:10.1152/ajpendo.00362.2014
38. Simoneau, JA, Veerkamp, JH, Turcotte, LP, and Kelley, DE. Markers of capacity to utilize fatty acids in human skeletal muscle: relation to insulin resistance and obesity and effects of weight loss. FASEB J (1999) 13(14):2051–60. doi:10.1096/fasebj.13.14.2051
39. van Dierendonck, X, de la Rosa Rodriguez, MA, Georgiadi, A, Mattijssen, F, Dijk, W, van Weeghel, M, et al. HILPDA uncouples lipid droplet accumulation in adipose tissue macrophages from inflammation and metabolic dysregulation. Cell Rep (2020) 30(6):1811–22.e6. doi:10.1016/j.celrep.2020.01.046
40. Morigny, P, Houssier, M, Mouisel, E, and Langin, D. Adipocyte lipolysis and insulin resistance. Biochimie (2016) 125:259–66. doi:10.1016/j.biochi.2015.10.024
41. Xu, X, Grijalva, A, Skowronski, A, van Eijk, M, Serlie, MJ, and Ferrante, AW. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab (2013) 18(6):816–30. doi:10.1016/j.cmet.2013.11.001
42. Kratz, M, Coats, BR, Hisert, KB, Hagman, D, Mutskov, V, Peris, E, et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab (2014) 20(4):614–25. doi:10.1016/j.cmet.2014.08.010
43. Prieur, X, Mok, CY, Velagapudi, VR, Nunez, V, Fuentes, L, Montaner, D, et al. Differential lipid partitioning between adipocytes and tissue macrophages modulates macrophage lipotoxicity and M2/M1 polarization in obese mice. Diabetes (2011) 60(3):797–809. doi:10.2337/db10-0705
44. Koonen, DP, Glatz, JF, Bonen, A, and Luiken, JJ. Long-chain fatty acid uptake and FAT/CD36 translocation in heart and skeletal muscle. Biochim Biophys Acta (Bba) - Mol Cel Biol Lipids (2005) 1736(3):163–80. doi:10.1016/j.bbalip.2005.08.018
45. Coburn, CT, Knapp, FF, Febbraio, M, Beets, AL, Silverstein, RL, and Abumrad, NA. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J Biol Chem (2000) 275(42):32523–9. doi:10.1074/jbc.m003826200
46. Zhou, J, Febbraio, M, Wada, T, Zhai, Y, Kuruba, R, He, J, et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARγ in promoting steatosis. Gastroenterology (2008) 134(2):556–67.e1. doi:10.1053/j.gastro.2007.11.037
47. Silverstein, RL, and Febbraio, M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal (2009) 2(72):re3. doi:10.1126/scisignal.272re3
48. Stewart, CR, Stuart, LM, Wilkinson, K, van Gils, JM, Deng, J, Halle, A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol (2010) 11(2):155–61. doi:10.1038/ni.1836
49. Hoebe, K, Georgel, P, Rutschmann, S, Du, X, Mudd, S, Crozat, K, et al. CD36 is a sensor of diacylglycerides. Nature (2005) 433(7025):523–7. doi:10.1038/nature03253
50. Nicholls, HT, Kowalski, G, Kennedy, DJ, Risis, S, Zaffino, LA, Watson, N, et al. Hematopoietic cell-restricted deletion of CD36 reduces high-fat diet-induced macrophage infiltration and improves insulin signaling in adipose tissue. Diabetes (2011) 60(4):1100–10. doi:10.2337/db10-1353
51. Lee, YS, Kim, JW, Osborne, O, Oh, DY, Sasik, R, Schenk, S, et al. Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell (2014) 157(6):1339–52. doi:10.1016/j.cell.2014.05.012
52. Chen, Y, Yang, M, Huang, W, Chen, W, Zhao, Y, Schulte, ML, et al. Mitochondrial metabolic reprogramming by CD36 signaling drives macrophage inflammatory responses. Circ Res (2019) 125(12):1087–102. doi:10.1161/circresaha.119.315833
53. Murthy, M, and Pande, S. Mechanism of carnitine acylcarnitine translocase-catalyzed import of acylcarnitines into mitochondria. J Biol Chem (1984) 259(14):9082–9. doi:10.1016/s0021-9258(17)47268-1
54. McGarry, JD, and Brown, NF. The mitochondrial carnitine palmitoyltransferase system—from concept to molecular analysis. Eur J Biochem (1997) 244(1):1–14. doi:10.1111/j.1432-1033.1997.00001.x
55. Doenst, T, Nguyen, TD, and Abel, ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res (2013) 113(6):709–24. doi:10.1161/circresaha.113.300376
56. Savage, DB, Choi, CS, Samuel, VT, Liu, Z-X, Zhang, D, Wang, A, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J Clin Invest (2006) 116(3):817–24. doi:10.1172/jci27300
57. Wakil, SJ, and Abu-Elheiga, LA. Fatty acid metabolism: target for metabolic syndrome. J lipid Res (2009) 50:S138–S143. doi:10.1194/jlr.r800079-jlr200
58. Dyck, JR, and Lopaschuk, GD. Malonyl CoA control of fatty acid oxidation in the ischemic heart. J Mol Cell Cardiol (2002) 34(9):1099–109. doi:10.1016/s0022-2828(02)92060-2
59. Ussher, JR, and Lopaschuk, GD. The malonyl CoA axis as a potential target for treating ischaemic heart disease. Cardiovasc Res (2008) 79(2):259–68. doi:10.1093/cvr/cvn130
60. Stuve, P, Minarrieta, L, Erdmann, H, Arnold-Schrauf, C, Swallow, M, Guderian, M, et al. De novo fatty acid synthesis during mycobacterial infection is a prerequisite for the function of highly proliferative T cells, but not for dendritic cells or macrophages. Front Immunol (2018) 9:495. doi:10.3389/fimmu.2018.00495
61. Yeudall, S, Upchurch, CM, Seegren, PV, Pavelec, CM, Greulich, J, Lemke, MC, et al. Macrophage acetyl-CoA carboxylase regulates acute inflammation through control of glucose and lipid metabolism. Sci Adv (2022) 8(47):eabq1984. doi:10.1126/sciadv.abq1984
62. Namgaladze, D, and Brune, B. Macrophage fatty acid oxidation and its roles in macrophage polarization and fatty acid-induced inflammation. Biochim Biophys Acta (Bba) - Mol Cell Biol Lipids (2016) 1861(11):1796–807. doi:10.1016/j.bbalip.2016.09.002
63. Camell, CD, Nguyen, KY, Jurczak, MJ, Christian, BE, Shulman, GI, Shadel, GS, et al. Macrophage-specific de novo synthesis of ceramide is dispensable for inflammasome-driven inflammation and insulin resistance in obesity. J Biol Chem (2015) 290(49):29402–13. doi:10.1074/jbc.m115.680199
64. Morgan, PK, Huynh, K, Pernes, G, Miotto, PM, Mellett, NA, Giles, C, et al. Macrophage polarization state affects lipid composition and the channeling of exogenous fatty acids into endogenous lipid pools. J Biol Chem (2021) 297(6):101341. doi:10.1016/j.jbc.2021.101341
65. Prieur, X, Rőszer, T, and Ricote, M. Lipotoxicity in macrophages: evidence from diseases associated with the metabolic syndrome. Biochim Biophys Acta (BBA)-Molecular Cell Biol Lipids (2010) 1801(3):327–37. doi:10.1016/j.bbalip.2009.09.017
66. Namgaladze, D, Lips, S, Leiker, TJ, Murphy, RC, Ekroos, K, Ferreiros, N, et al. Inhibition of macrophage fatty acid β-oxidation exacerbates palmitate-induced inflammatory and endoplasmic reticulum stress responses. Diabetologia (2014) 57(5):1067–77. doi:10.1007/s00125-014-3173-4
67. Holland, WL, Bikman, BT, Wang, LP, Yuguang, G, Sargent, KM, Bulchand, S, et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest (2011) 121(5):1858–70. doi:10.1172/jci43378
68. Vandanmagsar, B, Youm, YH, Ravussin, A, Galgani, JE, Stadler, K, Mynatt, RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med (2011) 17(2):179–88. doi:10.1038/nm.2279
69. Suganami, T, Nishida, J, and Ogawa, Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: role of free fatty acids and tumor necrosis factor alpha. Arteriosclerosis, Thromb Vasc Biol (2005) 25(10):2062–8. doi:10.1161/01.atv.0000183883.72263.13
70. Nakarai, H, Yamashita, A, Nagayasu, S, Iwashita, M, Kumamoto, S, Ohyama, H, et al. Adipocyte-macrophage interaction may mediate LPS-induced low-grade inflammation: potential link with metabolic complications. Innate Immun (2012) 18(1):164–70. doi:10.1177/1753425910393370
71. Nguyen, MT, Favelyukis, S, Nguyen, AK, Reichart, D, Scott, PA, Jenn, A, et al. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem (2007) 282(48):35279–92. doi:10.1074/jbc.m706762200
72. Lancaster, GI, Langley, KG, Berglund, NA, Kammoun, HL, Reibe, S, Estevez, E, et al. Evidence that TLR4 is not a receptor for saturated fatty acids but mediates lipid-induced inflammation by reprogramming macrophage metabolism. Cell Metab (2018) 27(5):1096–110.e5. doi:10.1016/j.cmet.2018.03.014
73. Dahik, VD, Frisdal, E, and Le Goff, W. Rewiring of lipid metabolism in adipose tissue macrophages in obesity: impact on insulin resistance and type 2 diabetes. Int J Mol Sci (2020) 21(15):5505. doi:10.3390/ijms21155505
74. Shi, H, Kokoeva, MV, Inouye, K, Tzameli, I, Yin, H, and Flier, JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest (2006) 116(11):3015–25. doi:10.1172/jci28898
75. Ham, M, Lee, JW, Choi, AH, Jang, H, Choi, G, Park, J, et al. Macrophage glucose-6-phosphate dehydrogenase stimulates proinflammatory responses with oxidative stress. Mol Cell Biol (2013) 33(12):2425–35. doi:10.1128/mcb.01260-12
76. Lee, JY, Sohn, KH, Rhee, SH, and Hwang, D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem (2001) 276(20):16683–9. doi:10.1074/jbc.m011695200
77. Schulze, PC, Drosatos, K, and Goldberg, IJ. Lipid use and misuse by the heart. Circ Res (2016) 118(11):1736–51. doi:10.1161/circresaha.116.306842
78. Karwi, QG, Ho, KL, Pherwani, S, Ketema, EB, Sun, Q, and Lopaschuk, GD. Concurrent diabetes and heart failure: interplay and novel therapeutic approaches. Cardiovasc Res (2022) 118(3):686–715. doi:10.1093/cvr/cvab120
79. Heather, LC, Gopal, K, Srnic, N, and Ussher, JR. Redefining diabetic cardiomyopathy: perturbations in substrate metabolism at the heart of its pathology. Diabetes (2024) 73(5):659–70. doi:10.2337/dbi23-0019
80. Boehm, EA, Jones, BE, Radda, GK, Veech, RL, and Clarke, K. Increased uncoupling proteins and decreased efficiency in palmitate-perfused hyperthyroid rat heart. Am J Physiology-Heart Circulatory Physiol (2001) 280(3):H977–83. doi:10.1152/ajpheart.2001.280.3.h977
81. Alavaikko, M, Elfving, R, Hirvonen, J, and Jarvi, J. Triglycerides, cholesterol, and phospholipids in normal heart papillary muscle and in patients suffering from diabetes, cholelithiasis, hypertension, and coronary atheroma. J Clin Pathol (1973) 26(4):285–93. doi:10.1136/jcp.26.4.285
82. Rijzewijk, LJ, van der Meer, RW, Smit, JW, Diamant, M, Bax, JJ, Hammer, S, et al. Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. J Am Coll Cardiol (2008) 52(22):1793–9. doi:10.1016/j.jacc.2008.07.062
83. Chokshi, A, Drosatos, K, Cheema, FH, Ji, R, Khawaja, T, Yu, S, et al. Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation (2012) 125(23):2844–53. doi:10.1161/circulationaha.111.060889
84. Alvarez-Guardia, D, Palomer, X, Coll, T, Serrano, L, Rodriguez-Calvo, R, Davidson, MM, et al. PPARβ/δ activation blocks lipid-induced inflammatory pathways in mouse heart and human cardiac cells. Biochim Biophys Acta (Bba) - Mol Cell Biol Lipids (2011) 1811(2):59–67. doi:10.1016/j.bbalip.2010.11.002
85. Puchalska, P, Martin, SE, Huang, X, Lengfeld, JE, Daniel, B, Graham, MJ, et al. Hepatocyte-macrophage acetoacetate shuttle protects against tissue fibrosis. Cell Metab (2019) 29(2):383–98.e7. doi:10.1016/j.cmet.2018.10.015
86. Huang, C, Wang, J, Liu, H, Huang, R, Yan, X, Song, M, et al. Ketone body β-hydroxybutyrate ameliorates colitis by promoting M2 macrophage polarization through the STAT6-dependent signaling pathway. BMC Med (2022) 20(1):148. doi:10.1186/s12916-022-02352-x
87. Puchalska, P, and Crawford, PA. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab (2017) 25(2):262–84. doi:10.1016/j.cmet.2016.12.022
88. Youm, YH, Nguyen, KY, Grant, RW, Goldberg, EL, Bodogai, M, Kim, D, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat Med (2015) 21(3):263–9. doi:10.1038/nm.3804
89. Jain, SK, Kannan, K, Lim, G, McVie, R, and Bocchini, JA. Hyperketonemia increases tumor necrosis factor-α secretion in cultured U937 monocytes and type 1 diabetic patients and is apparently mediated by oxidative stress and cAMP deficiency. Diabetes (2002) 51(7):2287–93. doi:10.2337/diabetes.51.7.2287
90. Kanikarla-Marie, P, and Jain, SK. Hyperketonemia (acetoacetate) upregulates NADPH oxidase 4 and elevates oxidative stress, ICAM-1, and monocyte adhesivity in endothelial cells. Cell Physiol Biochem (2015) 35(1):364–73. doi:10.1159/000369702
91. Kurepa, D, Pramanik, AK, Kakkilaya, V, Caldito, G, Groome, LJ, Bocchini, JA, et al. Elevated acetoacetate and monocyte chemotactic protein-1 levels in cord blood of infants of diabetic mothers. Neonatology (2012) 102(3):163–8. doi:10.1159/000339286
92. Rath, M, Müller, I, Kropf, P, Closs, EI, and Munder, M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol (2014) 5:532. doi:10.3389/fimmu.2014.00532
93. Peyssonnaux, C, Datta, V, Cramer, T, Doedens, A, Theodorakis, EA, Gallo, RL, et al. HIF-1α expression regulates the bactericidal capacity of phagocytes. J Clin Invest (2005) 115(7):1806–15. doi:10.1172/jci23865
94. Modolell, M, Corraliza, IM, Link, F, Soler, G, and Eichmann, K. Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH 1 and TH 2 cytokines. Eur J Immunol (1995) 25(4):1101–4. doi:10.1002/eji.1830250436
95. Ren, W, Xia, Y, Chen, S, Wu, G, Bazer, FW, Zhou, B, et al. Glutamine metabolism in macrophages: a novel target for obesity/type 2 diabetes. Adv Nutr (2019) 10(2):321–30. doi:10.1093/advances/nmy084
96. Jha, AK, Huang, SC, Sergushichev, A, Lampropoulou, V, Ivanova, Y, Loginicheva, E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity (2015) 42(3):419–30. doi:10.1016/j.immuni.2015.02.005
97. Liu, P-S, Wang, H, Li, X, Chao, T, Teav, T, Christen, S, et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol (2017) 18(9):985–94. doi:10.1038/ni.3796
98. Blanc, MC, Moinard, C, Béziel, A, Darquy, S, Cynober, L, and De Bandt, JP. Arginine and glutamine availability and macrophage functions in the obese insulin-resistant Zucker Rat. J Cell Physiol (2005) 202(1):153–9. doi:10.1002/jcp.20092
99. Costa Rosa, LFBP, Curi, R, Murphy, C, and Newsholme, P. Effect of adrenaline and phorbol myristate acetate or bacterial lipopolysaccharide on stimulation of pathways of macrophage glucose, glutamine and O2 metabolism. Evidence for cyclic AMP-dependent protein kinase mediated inhibition of glucose-6-phosphate dehydrogenase and activation of NADP+-dependent ‘malic’enzyme. Biochem J (1995) 310(2):709–14. doi:10.1042/bj3100709
100. Du, C, Liu, WJ, Yang, J, Zhao, SS, and Liu, HX. The role of branched-chain amino acids and branched-chain α-keto acid dehydrogenase kinase in metabolic disorders. Front Nutr (2022) 9:932670. doi:10.3389/fnut.2022.932670
101. Zhenyukh, O, Civantos, E, Ruiz-Ortega, M, Sanchez, MS, Vazquez, C, Peiro, C, et al. High concentration of branched-chain amino acids promotes oxidative stress, inflammation and migration of human peripheral blood mononuclear cells via mTORC1 activation. Free Radic Biol Med (2017) 104:165–77. doi:10.1016/j.freeradbiomed.2017.01.009
Keywords: macrophage, metabolism, obesity, type-2 diabetes, glucose, fatty acids, ketones, amino acids
Citation: Wong A, Sun Q, Latif II and Karwi QG (2024) Metabolic flux in macrophages in obesity and type-2 diabetes. J. Pharm. Pharm. Sci 27:13210. doi: 10.3389/jpps.2024.13210
Received: 30 April 2024; Accepted: 14 June 2024;
Published: 26 June 2024.
Edited by:
John Reyes Ussher, University of Alberta, CanadaCopyright © 2024 Wong, Sun, Latif and Karwi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qutuba G. Karwi, cXV0dWJhLmthcndpQG11bi5jYQ==