 Michał Rurek*
Michał Rurek* Mikołaj Smolibowski
Mikołaj Smolibowski- Department of Molecular and Cellular Biology, Institute of Molecular Biology and Biotechnology, Faculty of Biology, Adam Mickiewicz University, Poznań, Poland
Plant transcriptomes are complex entities shaped spatially and temporally by a multitude of stressors. The aim of this review was to summarize the most relevant transcriptomic responses to selected abiotic (UV radiation, chemical compounds, drought, suboptimal temperature) and biotic (bacteria, fungi, viruses, viroids) stress conditions in a variety of plant species, including model species, crops, and medicinal plants. Selected basic and applicative studies employing RNA-seq from various sequencing platforms and single-cell RNA-seq were involved. The transcriptomic responsiveness of various plant species and the diversity of affected gene families were discussed. Under stress acclimation, plant transcriptomes respond particularly dynamically. Stress response involved both distinct, but also similar gene families, depending on the species, tissue, and the quality and dosage of the stressor. We also noted the over-representation of transcriptomic data for some plant organs. Studies on plant transcriptomes allow for a better understanding of response strategies to environmental conditions. Functional analyses reveal the multitude of stress-affected genes as well as acclimatory mechanisms and suggest metabolome diversity, particularly among medicinal species. Extensive characterization of transcriptomic responses to stress would result in the development of new cultivars that would cope with stress more efficiently. These actions would include modern methodological tools, including advanced genetic engineering, as well as gene editing, especially for the expression of selected stress proteins in planta and for metabolic modifications that allow more efficient synthesis of secondary metabolites.
Introduction
Higher plants, known as vascular or telome plants (Thelomophyta), appeared during plant evolution back in the Palaeophytic era. They are characterized by the development of tissues that distribute water, mineral compounds, and photosynthesis products, and the dominance of the sporophyte (Kenrick and Crane, 1997; Forster et al., 2007). Due to the sessile life cycle, higher plants respond adequately to unfavorable conditions at multiple levels, including transcriptomic one (Morris et al., 2018).
The plant transcriptome is a complete pool of various RNA molecules (mRNA, rRNA, tRNA, as well as numerous ncRNAs) belonging to the translated fraction of the genome that responds to its environment (Imadi et al., 2015). Transcriptomics belongs to key “omics” studies that link genomic and proteomic “worlds” by analyses of RNA, a biopolymer with a central role in the transfer of genetic information and regulation of gene expression. Transcriptomics shows a more universal status than other “omics” disciplines. It offers complex and deep insights into studying gene expression in whole plants or plant organs/tissues; factors that regulate the transcriptome spatially and temporally can also be characterized (Alkan et al., 2011; Lowe et al., 2017; Zhang, 2019; Athanasopoulou et al., 2021). Moreover, plant genome assembly is more complex and expensive compared to RNA sequencing (RNA-seq), and when a reference genome is absent, the transcriptome can be used to assess plant overall transcriptional activity. Transcriptomics also allows for the quantification of low-abundance transcripts or their structural variants and estimation of the correlation of gene expression with biological traits. Additionally, the transcriptome outperforms the genome, allowing the characterization of genes related to therapeutic compound biogenesis (Wang et al., 2009). However, transcriptomics seems to be inappropriate for identifying genes with large impacts on adaptive responses to the environment due to a small number of genes with large impacts on fitness. When only transcriptomics is used to identify genes underlying environmental adaptations, constitutively expressed regulatory genes that play a major role in setting tolerance limits are often over-represented (Evans, 2015). Other disadvantages are serious challenges in analysing large datasets, as they demand a lot of bioinformatic tools, and most importantly, costs of sequencing (discussed below). The transcriptomic data may also contain noise enhanced by technical variations and batch effects resulting from inter-sample differences that were not rooted in the experimental design (Sprang et al., 2022).
Tissue-specific transcriptomics offer particularly valuable information on the underlying molecular processes that govern tissue-specific functions; furthermore, specific genes and regulatory mechanisms that display unique roles in diverse tissues can be better characterized (Booth et al., 2022). Droplet single cell RNA-seq (with most prominent platforms, including 10x Genomics) and spatial RNA-seq (with microdissection, spatial imaging and spatial coding approaches) allow the possibility of getting insight into the heterogeneity of tissue transcriptomes, to identify cell types and markers, and to analyse gene and regulatory networks under developmental and environmental factors. Their strengths include, for instance, the availability of spatial information and high resolution performance (Cervantes-Pérez et al., 2022; Chen et al., 2023; Wang et al., 2024).
The recent and prompt development of high-throughput RNA-seq platforms with a subsequent decrease in sequencing costs, as well as data meta-analyses, advanced on plant transcriptome studies (Tyagi et al., 2022). Currently, third-generation sequencing, including SMRT (single-molecule real-time) and Nanopore sequencing allows to obtain longer sequence reads, which challenged transcriptomic analyses. Long read sequencing is accurate and allows detection of alternative splicing events. SMRT sequencing employs sequencing by synthesis, linking of chemical groups to reduce background noise and is based on properties of zero-mode waveguides. In SMRT protocol, there is no need for amplification. SMRT sequencing was used by Pacific Biosciences of California (PacBio) platform. Nanopore platform is based on electrical signal sequencing and offers particularly long reads (Li et al., 2018; Ma L. N. et al., 2019; Huang et al., 2021). However, Illumina RNA-seq is still the most preferred sequencing platform in quantitative analyses (Supplementary Table S1).
The study of tissue-specific changes in gene expression under stress is instrumental in the development of strategies to improve plant response under environmental conditions (Berkowitz et al., 2021; Tyagi et al., 2022). In previous years, some reviews focused on methodological advances in plant transcriptomics (Schliesky et al., 2012; Tyagi et al., 2022; Chen et al., 2023; Wang et al., 2024). Species-specific omics analyses characterized the relevance of transcription factors (TFs), hormones, translational reprogramming and epigenetic level, as well as phenotypic and physiological levels in stress response (Singh et al., 2016; Ahmad, 2022; Bhat et al., 2022; Hu et al., 2022; Kourani et al., 2022; Son and Park, 2023; Tu et al., 2023). Some studies also focused on the roles of non-coding RNA in stress response (Yu et al., 2019; Jin et al., 2024). However, an updated review discussing transcriptomic responses to various stress conditions assayed by high-throughput approaches from various plant species is currently needed.
In this paper, the diversity of transcriptomic responses to various stressors, both abiotic (including chemical treatments, UV radiation, drought, cold and heat) and biotic (fungal, bacterial, and viral/viroid infections) ones, will be presented, and the results will be discussed in tissue/developmental and temporal contexts, reflecting the transcriptomic dynamicity. We will focus on current studies employing high-throughput analyses, for instance, RNA-seq from various experimental platforms, as well as microarrays. We will also summarise the transcriptomic responses of not only the model but also useful crop and medicinal species, which can be used for the development of future stress-resistant cultivars by genetic and metabolic engineering (Figure 1).
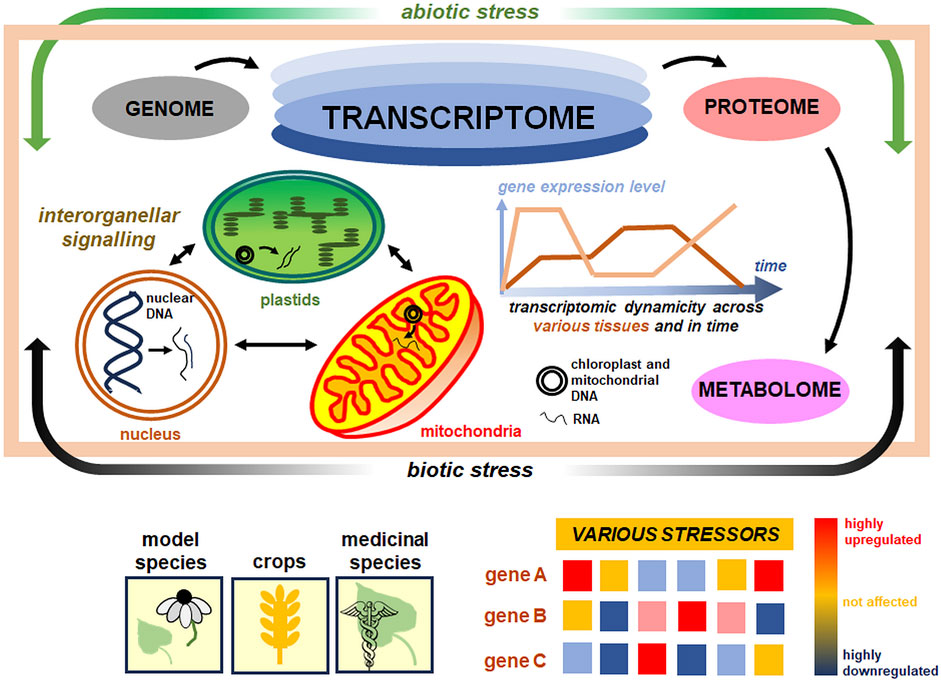
Figure 1. Plant transcriptome as a central stress-responsive entity in the cell. The plant transcriptome is a highly dynamic structure in plant cells (bluish ovals at the top). It links genomic, proteomic, and metabolomic levels, responding on a multidimensional scale, across various tissues, and along time (the center). The response of plant nuclear and organellar transcriptomes is also shaped by a number of factors, including abiotic and biotic stressors (green and black arrows encompassing the light brown central rectangle), which affect the differential expression pattern of various gene sets (bottom). For proper organellar biogenesis under stress acclimation, inter-organellar signaling between actively transcribed nuclear, plastid, and mitochondrial genomes is indispensable (small arrows within marked organelles on the panel in the center and to the left). The diversity of transcriptomes from model, crop, and medicinal species (bottom) under selected stress conditions was discussed in this paper. More details in the text.
Alterations in plant transcriptomes during abiotic stress
Stress conditions can be defined as internal and external cues that affect the efficiency of physiological, metabolic, and molecular plant processes, leading to a reduction in the efficiency of energy-to-biomass conversion. They can be divided into abiotic and biotic ones (Umar et al., 2021).
Abiotic stress results from the action of multiple physical or chemical stimuli (Gull et al., 2019; Wang et al., 2020). A comparison of the plethora of enriched functional terms representing numerous genes and transcription factors (TFs) responsive to various abiotic stressors from RNA-seq studies is shown in Figure 2 and additional quantitative details on genes affected by abiotic stress from high-throughput transcriptomic studies are also given in Supplementary Table S1, where details on stress treatments and the respective references to the literature are also shown. The experimental studies discussed in this review showed the huge variability of the gene response under those conditions, even between similar treatments. However, the stress response involves not only functional terms/ TFs common for all abiotic stressors discussed here (representing differentially expressed genes [DEGs] for photosynthetic genes or genes controlling secondary metabolite biosynthesis as well as MYB TFs), but also specific ones for each treatment; they were also presented in Venn diagrams, although depending on the stressor (Figure 2). Leaves, which are involved in the metabolism of carbon skeletons and the capture of photosynthetic energy, belong to plant organs particularly affected by unfavorable environmental conditions. However, studies on the impact of stress on leaf tissues, contrary to roots, are still underrepresented (Berkowitz et al., 2021).
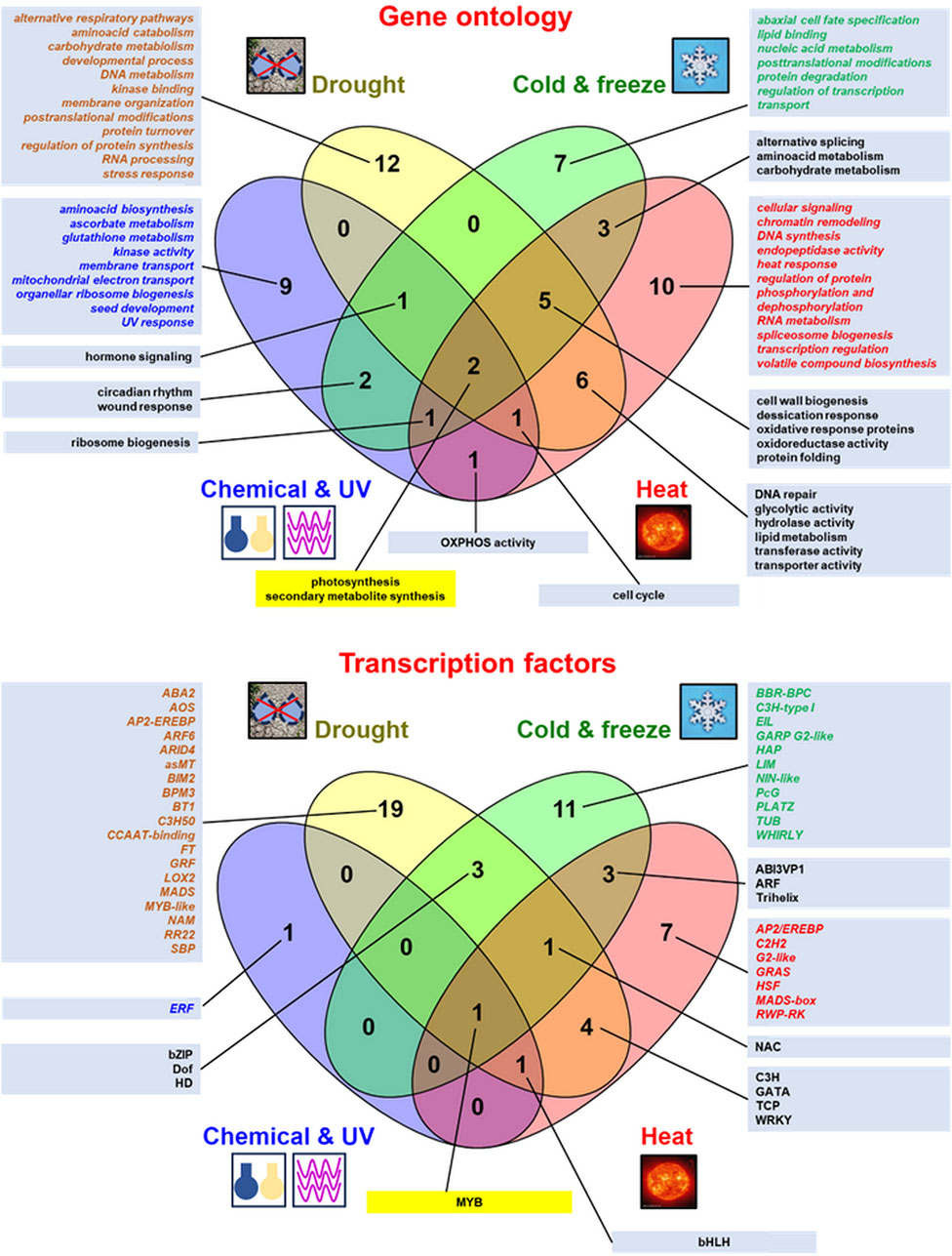
Figure 2. Comparison of the most relevant gene ontology (GO:) terms of plant genes and transcription factors active under abiotic stress conditions from various RNA-seq studies. The data were presented in Venn diagrams (drawn by Venny v. 2.1 from https://bioinfogp.cnb.csic.es/tools/venny/). The top enriched GO: terms (mostly relevant molecular functions and biological procesess) as well as transcription factors for regulated genes from the discussed studies were indicated. The data specific for the given stressor were denoted in italics and by different font colors (for the chemical treatment and UV radiation in blue, for drought in brown, for cold and freeze in green and for heat stress in red). GO: terms and transcription factors common for responses to all abiotic stressors were displayed within yellow text boxes. More details in the text.
UV radiation and chemical treatments
Variability of Arabidopsis leaf tissue transcriptomic responses under UV radiation, as well as under chemical treatments [e.g., antimycin A, 3-amino-1,2,4-triazole, methyl viologen, and salicylic acid (SA)] was characterized by Berkowitz et al. (2021). Responses of leaf tissues to these stressors were complex; for example, UV-affected genes were expressed mainly in the vasculature and epidermis. Treatment with 3-amino-1,2,4-triazole and SA downregulated genes for photosynthetic proteins in all tissues of Arabidopsis, while methylviologen upregulated the genes for the PS subunits, and UV radiation downregulated the photosynthetic genes in epidermis and upregulated them in mesophyll. Arabidopsis genes for oxidoreductase activity, porphyrin metabolism, plastid organization, and carbohydrate metabolism regulation were also affected by UV-B. Interestingly, Arabidopsis genes for proteins for chlorophyll biogenesis, protein folding, oxidoreductase and ligase genes, and glyceraldehyde-3-phosphate dehydrogenase were differentially regulated between UV-A and UV-B treatments. Tissues studied also showed distinct mitochondrial responses to antimycin A, which affected the expression pattern of respiratory genes, general oxidoreductase activity genes, glutathione transferase, as well as genes related to Ser/Thr kinase activity and membrane transport (Berkowitz et al., 2021).
Among DEGs upregulated in Vaccinium corymbosum, genes involved in plant hormone signal transduction were significantly enriched after 1 h, followed by genes involved in phenylpropanoid biosynthesis after 3 h, and genes involved in the flavonoid anthocyanin pathway after 6 h of exposure to UV-B. These results suggest that phytohormone-related genes contribute to the primary response to UV-B radiation. However, the highest number of DEGs appeared among V. corymbosum plants exposed to UV-B treatment for 24 h. Genes involved in proanthocyanidin and flavanol biosynthesis (PAL1, 4CL2, CHS, CHI3, VcFLS and VcUFGT) were upregulated by UV, and their expression level lasted a long time after 24 h of treatment (Song et al., 2022). These DEGs resembled Arabidopsis genes affected by radiation (Berkowitz et al., 2021). Common genes for UV-B response in Arabidopsis and Pachycladon cheesemanii included genes for amino acid, vitamin, pigment, and secondary compound metabolism (Dong, 2024).
The impact of UV-B radiation on the transcriptome of Glycyrrhiza uralensis, a potent medicinal species, was investigated by Zhang et al. (2018). Participation of amino acid metabolism and enzymes in secondary metabolite pathways in the response to radiation at five different time points was suggested. Genes for various amino acid metabolic pathways were differentially enriched depending on UV-B duration, however, DEGs for enzymes of cysteine and methionine metabolism were mainly enriched in all treatments.
Polygonum cuspidatum, a medicinal plant species used in traditional Chinese medicine, exhibits numerous medicinal applications (Ke et al., 2023). The impact of UV-C radiation on the transcriptome of Polygonum cuspidatum leaves was studied by Liu et al. (2019); more DEGs (including more downregulated genes) were involved in shorter radiation response. Furthermore, this was accompanied by an increased concentration of resveratrol (the most important stilbene phytoalexin polyphenolic compounds) in the leaves of P. cuspidatum. Resveratrol used in the nutraceutical industry exhibits anticancer and anti-ageing properties. Under UV-C radiation, upregulated DEGs coded enzymes involved in proanthocyanidin and flavanol biosynthesis, however, chalcone synthase (CHS) gene was downregulated. Furthermore, MYB, bHLH, and ERF TFs appeared to be potential regulators of resveratrol biosynthesis genes. These results will help to find more practical applications of resveratrol synthesis (Liu et al., 2019). Upregulation of PAL, C4H, 4CL, and STS enzymes by means of genome editing may be positively associated with the resveratrol levels (Hasan and Bae, 2017).
Under high light treatment (with the enhanced UV radiation) of ginseng (Panax ginseng), another medicinal species, affected DEGs in the leaf transcriptome were mostly involved in various metabolic pathways and in the stress response. Stress-responsive functions were enriched among 33% of the upregulated DEGs, and the high light intensity and/or ROS response was associated with another 6% of the affected DEGs (Jung et al., 2020).
Lettuce (Lactuca sativa) grown under greenhouse conditions usually contains a lover level of ascorbic acid (ASC), an essential antioxidant nutrient for human health, compared to field-grown plants. To investigate the effect of radiation on ASC level in plants, lettuce plants were treated with various UV-B doses. Numerous DEGs were identified within lowly and highly radiated plants (Zhou et al., 2023). It was suggested that the expression of MIOX (for myo-inositol oxygenase, a key enzyme in the myo-inositol pathway), APX, and MDHAR may contribute to the indirect increase in the level of ASC induced by UV-B radiation (Lorence et al., 2004).
In general, plant transcriptomic responses to UV and chemical compound treatments were specifically enriched in genes for aminoacid biosynthesis, ascorbate and glutathione metabolism, kinase activity, membrane transport, mitochondrial electron transport proteins, organellar ribosome biogenesis and seed development (Zhang et al., 2018; Liu et al., 2019; Berkowitz et al., 2021; Song et al., 2022; Zhou et al., 2023; Dong, 2024). Interestingly, as shown on the Venn diagram, ERF proteins belong to TFs active under chemical treatment and UV radiation (Figure 2).
Water deficiency (drought)
As drought belongs to factors that affect the largest part of crop productivity, the analysis of multiple molecular responses by omics studies would allow characterising the mechanisms of drought in crops that result in the search for stress-resistant cultivars (Verma et al., 2013; Shanker et al., 2014).
Analysis of sweet potato (Ipomoea batatas) transcriptome in drought allowed identification of various upregulated genes for ABA, ethylene, and JA biosynthesis, indicating the relevance of hormonal signaling in a water deficit. Genes for ABI phosphatase and Ca2+-ATPase were severely altered, while genes for SA synthesis appeared not affected (Zhu et al., 2019). Also in chickpea (Cicer arietinum) numerous genes for AP2-EREBP, bHLH, bZIP, C3H, MYB, WRKY or MADS TFs that regulate signaling regulation, secondary metabolism, or transition to the generative phase were involved in drought acclimation (Kumar et al., 2019). The transcriptomic response of Phoebe bournei, a Chinese wood species, to drought also used DEGs for plant hormone signal transduction in addition to genes for redox homeostasis (POD, SOD, and CAT), phenylpropanoid, flavonoid and porphyrin biosynthesis, starch and sucrose metabolism, chlorophyll a/b binding proteins, and genes for numerous TFs from 25 families (Li et al., 2022).
Recent investigation of transcriptomes of two rice (Oryza sativa) cultivars that varied with stress resistance revealed that genes for hormone signaling (in line with Kumar et al., 2019; Zhu et al., 2019; Li et al., 2022 studies), LEA proteins, proteins related to redox homeostasis and NAC and ZIP TFs played crucial roles in developing drought tolerance (Tyagi et al., 2023). Additionally, in the transcriptome of Medicago falcata seedlings, DEGs for hormone signaling (ABA biosynthesis, JA biosynthesis), nucleic acid helicases, and diverse genes for RNA polymerases and DNA repair proteins were enriched. In contrast, gibberellin biogenesis genes were antagonistically expressed compared to ABA-related genes (except for the GID1 gene). Numerous TFs were also affected (Miao et al., 2015).
In Ceratostigma plantagineum, a resurrection species, studied by Xu et al. (2021), affected DEGs encoded proteins also active in hormone signaling, and in photosynthesis, stress response, amino acid catabolism, sucrose and fatty acid biogenesis, RNA processing and regulation, energy metabolism (distinctive in mild drought), protein modification and transport, and membrane organisation. Those data indicate the flexibility of primary and secondary metabolism in water shortage and re-watering, using, among others, an alternative respiratory pathway, the C3-CAM switch, and the GABA shunt. During global reanalysis of the Glycine max transcriptome, DEGs for proteins for hormone signaling, cell division, cell cycle, cell wall organization, stress responses, signal transduction, and regulation of gene expression, were regulated by progressing drought (De Oliveira-Busatto et al., 2022).
Mild drought-affected Arabidopsis genes code proteins that participate in ABA signaling, ROS biogenesis, response to osmotic stress, and also in cell wall remodelling and cell growth, among which multiple genes were previously not associated with drought-responsive mechanisms. Hormone signaling genes for PYRABACTIN RESISTANCE/ ABA receptors, two ACC oxidases and four ethylene response factors were downregulated and protein phosphatases 2C, HAB proteins, some ABA-responsive element-binding factors as well as some their target genes were all upregulated. Cell wall-loosening expansins, pectin lyases were also upregulated (Clauw et al., 2015). These data, which allowed insight into the transcriptomic landscape of six Arabidopsis accessions in drought, were further re-analysed by Benny et al. (2019), who underlined the importance of hydrogen peroxide, water deprivation, salinity, osmotic stress, and ABA-responsive proteins among upregulated genes. Transcriptomic analysis of rapeseed (Brassica napus), another representative of Brassicaceae, revealed that upregulated DEGs were related to the response to water deprivation, ABA signaling, osmotic stress, and other abiotic stimuli and lipid metabolism, as well as cutin, suberin, and wax biogenesis, fatty acid degradation, and secondary compound metabolism (Fang et al., 2022).
The multitude of various TFs was associated with the response to drought of Dendrobium sinense, an endemic species (Zhang et al., 2021). DEGs coded proteins for carbohydrate derivative and nucleotide binding, ATPase and oxidoreductase activity, pectin metabolism, and multiple TFs. Interestingly, more DEGs participated in a mild drought response, where detrimental downregulation prevailed (Zhang et al. (2021). Furthermore; Xia et al. (2024) broadened the analysis of Dendrobium drought responses by three additional species; the highest count of DEGs appeared in D. fimbriatum. Multiple DEGs among various Dendrobium species were involved in carbon metabolism and anthocyanin biosynthesis. Noticeable differences in the expression level of the PEPC gene (for phosphoenolpyruvate carboxylase) were associated with CAM and improved drought tolerance.
Artemisia annua, a medicinal species, is a potent worldwide source of artemisinin, an antimalarial compound. Attempts have been made to significantly increase artemisinin yield, and stress tolerance engineering would be one of such strategies. Drought response of A. annua leaf transcriptome employed many DEGs, including those coding for Δ-1-pyrroline-5-carboxylate synthetase, aquaporins, glyceraldehyde-3-phosphate dehydrogenase, LEA proteins, HSPs, glyoxalase I, glutathione-S-transferase, PR proteins, Ca2+-dependent protein kinases, as well as proteins involved in ethylene and oxylipin biosynthesis as well as NAC and MYB-related TFs (Vashisth et al., 2018).
Transcriptomes of two wheat (Triticum aestivum) varieties with contrasting drought resistance were compared; in stress resistant cultivars, the drought response involved genes for the synthesis of secondary metabolites and important transcription coregulators and TFs (Kumar et al., 2018).
Growth regulator 5-aminolevulinic acid (ALA) has been used to alleviate drought in grapevine (Vitis vinifera), by increasing antioxidative responses (Yang et al., 2023). Chlorophyll metabolism and photosynthetic apparatus were primarily affected by ALA, which uses synergistic mechanisms to alleviate drought. In the presence of ALA, alterations in the expression pattern of DEGs for chlorophyll biogenesis and Rubisco-related genes played an important role that allowed ALA to maintain cell homeostasis under water scarcity.
Little was known about the combined action of drought and cold on the plant transcriptome. Sharma et al. (2018) provided a comparative study of the impact of both stressors on the Arabidopsis transcriptome by meta-analysis of publicly available transcriptomic data. Responsive DEGs encoded proteins related to photosynthesis, respiratory burst, hormone response, signal transduction, and water deprivation, as well as some stress-specific genes. Furthermore, at least 43 diverse TFs were expressed in both treatments. Coolen et al. (2016) analysed Arabidopsis plants under drought combined with biotic treatments. The water deficit alone increased expression level of DEGs coding for proteins responding to oxygen-containing compounds and cell wall biogenesis. As the drought progressed, the more pronounced were the alterations in gene profiles. Each of the stressors induced specific expression profiles over time. In sequential stress application, Arabidopsis displayed transcriptome profiles similar to those of the second treatment, regardless of the nature of the first stressor. Overall, this study highlights the importance of stress signatures in identifying key molecular responses that act between various response pathways.
In general, hormone signaling pathways belong to the common terms for UV treatment (as discussed above; Figure 2; Song et al., 2022) and drought (Figure 2; Miao et al., 2015; Sharma et al., 2018; Kumar et al., 2019; Zhu et al., 2019; Fang et al., 2022; Li et al., 2022). The transcriptomic response in drought specifically affects a multitude of genes involved in alternative respiratory pathways, aminoacid catabolism and carbohydrate metabolism, developmental processes, DNA metabolism, kinase binding, membrane organisation, postranslational modifications, protein turnover, regulation of protein synthesis, RNA processing, and stress response. It is also specifically regulated by a particularly broad variety of TFs and transcriptional coactivators of various families (Figure 2; Kumar et al., 2018; Kumar et al., 2019; Benny et al., 2019; Zhu et al., 2019; Xu et al., 2021; De Oliveira-Busatto et al., 2022; Yang et al., 2023; Xia et al., 2024).
Elevated temperature (heat stress)
Elevated temperature affects cereal productivity, particularly male generative organ development and pollen maturation and viability (Young et al., 2004; Asseng et al., 2011; Wu et al., 2015). Heat stress leads to an increase in the level of reactive oxygen species (ROS) and a simultaneous decrease in ROS scavenger activity, leading to biomolecul damage and apoptosis (Bita and Gerats, 2013; Guan et al., 2013). Chrysanthemum leaf transcriptomes were analyzed in heat with or without melatonin (to alleviate the consequences of heat treatment; Xing et al., 2021). Heat alone resulted in massive downregulation of DEGs. In contrast, heat combined with melatonin increased expression level of several DEGs. Melatonin affected HSF and HSP, starch and sucrose metabolism, cell signaling, chlorophyll, flavonoid, carotenoid biosynthesis genes, and genes for various TFs.
Comparison of microspore transcriptomes under heat in two tomato (Solanum lycopersicum) cultivars with contrasting stress tolerance revealed among upregulated DEGs at least 11 HSP genes. Increased expression of the HSP and APX genes pinpoints the key role of antioxidant enzymes in the heat response (Frank et al., 2009). Valdés-López et al. (2016) studied dynamics of root hair transcriptome in soybean (G. max) subjected to heat at various time points. Responsive genes were classified into 10 functional modules regulated by a few TFs. In general, heat affected the expression pattern of DEGs for protein folding genes, but also for genes coding proteins for chromatin remodeling, and lipid and ATP synthesis, indicating for the importance of controlling water/nutrient intake by roots and the relevance of maintaining high ATP level under heat response.
Rice leaf transcriptome under thermal shift was studied by Rashid et al. (2020). Multiple genes were affected for the abiotic stress response and metabolite biosynthesis. Among the DEGs affected, only a few photosynthetic/ OXPHOS genes as well as some genes for glycolytic enzymes were present. Chen and Li (2017) investigated Brachypodium distachyon leaf transcriptome in heat. Affected DEGs coded proteins that participate in alternative RNA splicing, spliceosome, and PS biogenesis, indicating an increased extent of such events in response to high temperature in order to synthesize protein isoforms alleviating heat detrimental effects.
To describe thermotolerance and protective mechanisms against thermal stress in desert species, the Rhizya stricta transcriptome, the evergreen shrub, was analysed at elevated temperature. Upregulated genes coded HSPs, chaperones, UDP-glycosyltransferase, aquaporins and transparent protein testa 12, suggesting the distinctness of thermotolerance in leaves of Rhizya stricta, which is controlled primarily by improving protein folding and preventing protein degradation (Obaid et al., 2016). HSP genes and genes for flavonoid biosynthesis were upregulated in leaves of three tea cultivars (Camellia sinensis), important medicinal species, under heat (Huang et al., 2024). Studied cultivars differed in stress tolerance and exhibited mainly upregulated DEGs under heat acclimation, however, among the affected DEGs in all cultivars, the genes for photosynthetic activity were the most notable. In heat-tolerant cultivars at elevated temperatures, genes for proteins containing the chaperone domain, including universal stress proteins (USPs), small heat shock protein sHSP18.1, chaperonin-like protein 2 (CLP2), and the LEA5 protein were preferentially expressed. Additionally, the level of flavonols increased in heat-tolerant varieties, accompanied by increased expression of FLS genes. Therefore, in accordance with Obaid et al. (2016), Xing et al. (2021) and Frank et al. (2009) reports, the study by Huang et al. (2024) highlights the importance of chaperones and secondary metabolism in the heat response.
Heat stress often acts simultaneously with water deficit. Mikołajczak et al. (2023) focused on investigating the impact of heat stress, drought, and their joining effects on transcriptome of barley (Hordeum vulgare) flag leaves. In medium-sized leaves, short heat stress (similarly to drought) affected multiple genes, regardless of the duration of the stress. However, under longer heat and drought, more DEGs were affected in large leaves. Investigated stressors affected mainly the LEA and HSP genes. Overall, Mikołajczak et al. (2023) provided novel data on the molecular mechanisms of barley flag leaf that determine the response to drought and heat. Furthermore, according to Mahalingam et al. (2022), the number of DEGs increased in barley heads in the stress-tolerant genotype as heat progressed. Heat response involved genes for transporter proteins, and ABA response, and resulted in differential expression of LEA genes in stress-sensitive genotype. In contrast, genes for nonspecific lipid transfer proteins and carbonate dehydratase were enriched in a stress-tolerant cultivar. Heat with drought resulted in a notable increase in DEG number only in the stress-sensitive cultivar. In particular, at least 900 TFs controlled transcriptional reprogramming in two barley cultivars in all treatments.
Cellular signalling, including hormone signaling, is particularly important in multiple treatments, when heat is combined with other stressors. Martin et al. (2021) pointed out the broadening range of DEGs affected by double treatment (heat and drought) of Lolium temulentum, which encoded proteins for cell signaling, cell cycle, organellar biogenesis, binding, transport, oxidoreductase and antioxidative activit, as well as chaperones and multiple TFs. When heat stress acted together with an elevated level of CO2, detrimental effects were only partially alleviated by deregulation of primary and secondary metabolism genes in flag leaves of durum wheat (Triticum durum) flag leaves. Most affected DEGs coded proteins involved also in cellular signaling but also in stress response and nucleic acid metabolism. In particular, genes upregulated by CO2 were often downregulated by heat (they coded, among others, photosynthetic and OXPHOS proteins, proteins for hormone signaling, enzymes of lipid and amino acid metabolism and the glutathione-ascorbate cycle, nucleic acid metabolism, and transport proteins (Vicente et al., 2019). Suzuki et al. (2016) investigated the impact of heat and salinity on Arabidopsis transcriptome. DEGs regulated by heat and salt stress were enriched in genes coded proteins for ABA signaling, stress response, developmental processes, protein metabolism, and DNA-dependent transcription. However, in heat, DEGs for ABA-responsive proteins, glyoxylase 17 and catalase, among others, appeared to be responsive, indicating the importance of the antioxidative response.
Arabidopsis leaf transcriptome under various stress conditions (salinity, osmotic stress, and heat) was also investigated by Sewelam et al. (2020). Of all these treatments, elevated temperature appeared to have the most notable effect on transcriptomic profiles. DEGs affected by heat covered induction of eleven HSP genes, late embryogenesis abundant (LEA) proteins, receptor-like kinases (RLKs), glutathione S-transferases, genes for carbohydrate binding, UDP-galactosyltransferases, membrane transporters, and programmed cell death (PCD), and genes for WRKY TFs. However, heat treatment repressed several cell cycle genes, ribosomal protein genes, and genes involved in DNA synthesis and repair. In particular, osmotic stress and heat acted antagonistically, while double treatment largely reprogrammed the gene expression pattern. Heat in combination with salinity and osmotic stress also induced numerous mitochondrial genes, presumably as a compensatory response to excessive protein degradation (Rurek et al., 2018).
In general, multiple stress treatment (including heat) in plant transcriptomes results in different and distinct transcriptomic responses under single treatments. In particular, although drought often accompanies heat, those stressors result in the upregulation of different gene sets in various plant species (Mahalingam et al., 2022; Mikołajczak et al., 2023). The transcriptomic response to heat stress specifically engages DEGs for cellular signaling (particularly in multiple stress treatments and under melatonin supplementation; Suzuki et al., 2016; Vicente et al., 2019; Martin et al., 2021; Xing et al., 2021), and also genes for chromatin remodelling, DNA synthesis, endopeptidase activity, heat response, regulation of protein phosphorylation and dephosphorylation, RNA metabolism, spliceosome biogenesis, transcription regulation, volatile compound biosynthesis as well as AP2/EREBP, C2H2, G2-like, GRAS, HSF, MADS-box and RWP-RK TFs (Figure 2; Frank et al., 2009; Obaid et al., 2016; Suzuki et al., 2016; Valdés-López et al., 2016; Chen and Li, 2017; Rurek et al., 2018; Vicente et al., 2019; Rashid et al., 2020; Sewelam et al., 2020; Martin et al., 2021; Xing et al., 2021; Mahalingam et al., 2022; Mikołajczak et al., 2023; Huang et al., 2024).
Low temperature (cold, freeze)
Similarly to heat, also cold treatment and freeze result in plant growth and development aberrations, as well as direct inhibition of metabolic reactions. Due to the limited osmosis, cell dehydration and oxidative stress occur simultaneously with other detrimental responses. Most plants can gain tolerance to ice formation by gradually being exposed to reduced (non-freezing) temperatures under cold acclimation (Chinnusamy et al., 2007). In general, genes for the early cold response encoded a wide variety of TFs that regulate other gene expression (Gull et al., 2019).
Signal transduction and hormone signaling appeared also to be important for the low-temperature response. Amur vine (Vitis amurensis) transcriptomic response under cold used DEGs that encode proteins for signal transduction, transcription regulation, and alternative splicing. At least 38 major families of TFs (with 326 genes for TFs) involved in the regulation of cold response were detected, including several previously uncharacterized TF families homologous to Arabidopsis proteins (e.g., HAP2, ABI3VP1, ARF, PLATZ, LIM, atypical HD and MYB factors, BBR-BPC, zinc Dof, C3H-type I, EIL, GARP G2-like and Trihelix). Genes for CBL-interacting protein kinases participating in signal transduction were upregulated (Xu et al., 2014). Du et al. (2017) analysed the cold response of Agropyron mongolicum ABA receptors and upregulated genes for bZIP and NAC TFs. Most DEGs participated in carbohydrate metabolism, hormonal and phosphatidylinositol signaling, as well as biogenesis of numerous secondary metabolites. Also, the cold response of M. falcata focused on phytohormone and nodulation signaling, revealing some similarities with the drought replies; however, ABF1, GID1, and AUX genes were downregulated. Interestingly, GH3 auxin-responsive gene was extensively upregulated in cold (similarly to DIMI1, but contrary to DIMI2 and DIMI3, all of which encode important nodulation factors). Furthermore, at least 16 genes for MYB and 12 genes for NAC TFs were induced by cold, indicating their participation in cold tolerance (Miao et al., 2015). The transcriptomic response to cold in two rape cultivars (Brassica rapa) cultivars, varying with stress tolerance, also involved DEGs coding proteins for plant hormone signal transduction (MAPK signaling pathway) and also for photosynthesis, phenylpropanoid biosynthesis, lipid binding, plant-type cell wall, positive regulation of circadian rhythm, abaxial cell fate specification and basal TFs (Ma L. et al., 2019).
Arabidopsis chilling response covered almost half of expressed genes (Calixto et al., 2018; Liu et al., 2022). Cold response also involved genes for plant hormone signaling, but also for glucosinolate biosynthesis, transporter proteins, long ncRNA, RNA splicing, and spliceosome biogenesis, as well as multiple TFs from 52 families. Cold downregulated photosynthetic genes and short cold-induced quickly responding DEGs coding proteins for chloroplast organisation, ribosome biogenesis, and rRNA processing, which may activate cold tolerance. On the contrary, long cold affected DEGs for the cell response to hypoxia, fatty acid and flavonoid biosynthesis, as well as redox homeostasis and diverse HSP genes (Liu et al., 2022). Massive alternative splicing events occurred in the first few hours of cold treatment, including mobilisation of cold-responsive TFs, splicing factors, and selected RNA-binding proteins (Calixto et al., 2018). Interestingly, in P. cheesemanii, contrary to Arabidopsis, the cold response employed genes for glucosinolate metabolism. For Arabidopsis and P. cheesemanii, genes for wound-like circadian clock, as well as for secondary metabolite biogenesis, responded under the early cold response (Dong et al., 2023).
Cheng et al. (2019) described the effects of MeTCP4 (a specific cassava [Manihot esculenta] TF) overexpression in Arabidopsis plants during cold stress. Affected genes were classified as stress-responsive under all tested conditions, to DNA binding and TF activity in control, and to oxidoreductase, peroxidase, and antioxidative activity under cold treatment. Analysis of transcriptomic response of leaves of Lavandula angustifolia to cold revealed DEGs coding for photosynthetic proteins among affected genes. The most important functions of DEGs were also associated with the decreased stomatal conductance, ROS scavenging, and the development of cold tolerance. In general, these findings may allow for further engineering of cold tolerance in L. angustifolia to improve its medicinal potential, as this species is a source of aromatic compounds in traditional Chinese medicine (Li L. et al., 2023). In the leaf transcriptome of another medicinal species, A. annua, cold induced a multitude of genes for kinases, peroxidases, ABA biosynthesis, LEA and LEA-like proteins, various desaturases, glyoxalase I family protein, proteins for oxylipin and polyamine biosynthesis, Δ-1-pyrroline-5-carboxylate synthetase, NAC and MYB TFs (Vashisth et al., 2018).
Important metabolic regulations occur in plant organelles under cold acclimation. Naydenov et al. (2010) investigated the mitochondrial and nuclear transcriptomes of germinated wheat (T. aestivum). The upregulated genes encoded mitochondrial proteins, including Mn superoxide dismutase (SOD) and alternative oxidase (AOX); however, the level of expression of nuclear genes essential for mitochondrial biogenesis was visibly reduced. This indicates a fine-tuning of gene expression between mitochondrial and nuclear transcriptomes, executed by anterograde and retrograde signaling, affected by stress conditions.
On the whole, the cold response of plant transcriptomes specifically employs DEGs involved in abaxial cell fate specification, lipid binding, nucleic acid metabolism, post-translational modifications, protein degradation, regulation of transcription, and biomolecule transport. Cold response also specifically mobilizes a variety of TFs, including BBR-BPC, C3H-type I, EIL, GARP G2-like, HAP, LIM, NIN-like, PcG, PLATZ, TUB, and WHIRLY proteins (Figure 2; Naydenov et al., 2010; Xu et al., 2014; Miao et al., 2015; Du et al., 2017; Calixto et al., 2018; Vashisth et al., 2018; Cheng et al., 2019; Gull et al., 2019; Ma L. et al., 2019; Liu et al., 2022; Dong et al., 2023; Li L. et al., 2023). Hormone signalling is also affected by the cold response in numerous studies, including members of Fabaceae and Brassicaceae (Miao et al., 2015; Du et al., 2017; Vashisth et al., 2018; Gull et al., 2019; Ma L. et al., 2019; Liu et al., 2022).
Plant transcriptomic responses under biotic stress
Under global climate alterations, crop species are gradually exposed to increasing biotic stress. The findings discussed below may help develop plant cultivars that are highly resistant to fungal, bacterial and virus infections. To cope with biotic stress, plants have developed defensive responses precisely induced by pathogen attack (Verma et al., 2013). Plant cells contain plasma membrane receptors, which recognize pathogen-associated molecular patterns (PAMP). Subsequently, PAMP-triggered immunity (PTI) usually stops the infection before it spreads throughout the plant. Due to the constant combat between pathogens and their victims, pathogens can neutralize PTI by secreting special effector proteins into the cytosol. In response, plants developed the ability to detect microorganisms by effector-triggered immunity (ETI). Interactions between intracellular receptors designed to recognize effector molecules produced by pathogens and effectors trigger a complex network of cell responses to achieve infection resistance (McDowell and Dangl, 2000). To protect against pathogens, plants use an “oxidative outbreak” that initiates a hypersensitive response (HR) limiting the pathogen spread (Sato et al., 2010).
A comparison of the multitude of enriched functional terms representing numerous genes and transcription factors (TFs) that respond to various biotic stressors (fungal, bacterial, and virus/viroid infections) from RNA-seq studies is shown in Figure 3 and further quantitative details on genes affected by abiotic stress from high-throughput transcriptomic studies are also given in Supplementary Table S1, where details on various biotic treatments and references from the respective literature were given. Fungal, bacterial, and virus/ viroid infections involve common DEGs for carbohydrate metabolism, defence response, photosynthesis, protein phosphorylation, ribosome biogenesis, secondary metabolite biosynthesis, and transcription regulation. Biotic stress influence also expression patter of genes for hormone signaling/ signal transduction (more details below). All conditions of biotic stress mobilize WRKY factors responding to pathogen infection (Figure 3). Interestingly, terms for photosynthesis and secondary metabolite biosynthesis overlap with those for abiotic stress response; however, most functional terms and TFs in biotic infections differ from those of abiotic stressors (Figures 2, 3).
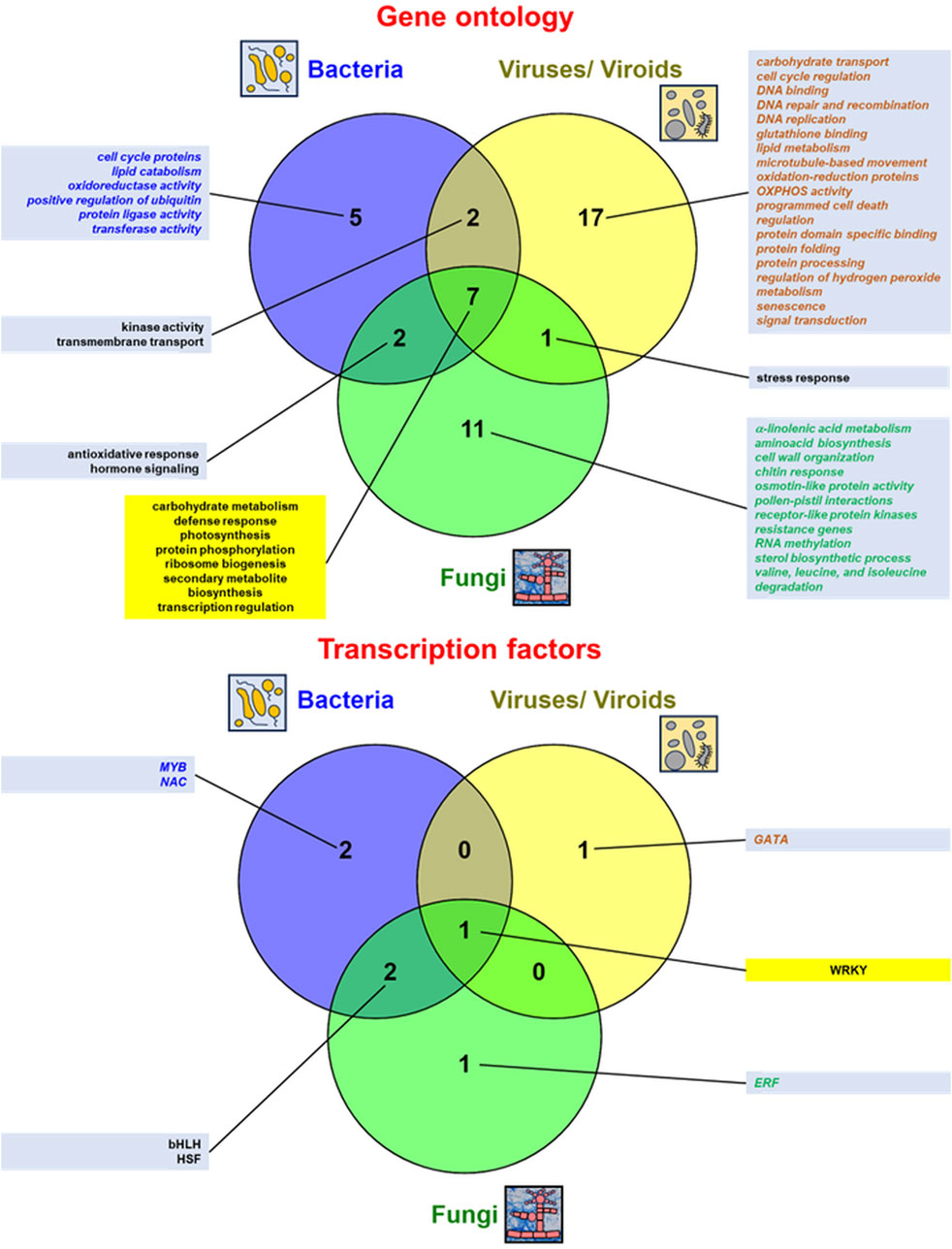
Figure 3. Comparison of the most relevant gene ontology (GO:) terms of plant genes and transcription factors active under biotic stress conditions from various RNA-seq studies. The data were presented in Venn diagrams (drawn by Venny v. 2.1 from https://bioinfogp.cnb.csic.es/tools/venny/). The top enriched GO: terms (mostly relevant molecular functions and biological procesess) as well as transcription factors for regulated genes from the discussed studies were indicated. The data specific for the given stressor were denoted in italics and by different font colors (for bacterial infections in blue, for virus/viroid infections in brown and for fungal infections in green). GO: terms and transcription factors common for responses to all abiotic stressors were displayed within yellow text boxes. More details in the text.
Fungal infections
Pathogenic fungi can be divided into biotrophic, necrotrophic, and hemibiotrophic species (biotrophic in their early stages and necrotrophic in their later stages of the life cycle). Analysis of transcriptome of pumpkin leaves (Cucurbita moschata) infected with powdery mildew (Blumeria graminis) 24 and 48 h after the infection onset showed a downregulation of multiple DEGs, including genes coding for resistance to powdery mildew and various TFs for ethylene signaling. Numerous host photosynthetic genes were also regulated. Upregulation of photosynthetic genes after 48 h of infection was associated with the appearance of initial fungal hyphae, which was also shown in infected wheat (T. aestivum) (Guo et al., 2018).
The transcriptomes of wheat leaves infected with powdery mildew (Erysiphales species) and striped rust (Puccinia striiformis) were analysed by Zhang et al. (2014). In powdery mildew infection, DEGs coded proteins for α-linolenic acid metabolism, as well as phenylpropanoid, flavonoid, phenylalanine, tyrosine and tryptophan biosynthesis. However, striped rust upregulated DEGs for photosynthetic proteins and proteins for ubiquinone biosynthesis. These results indicate the participation of different genes in response to various fungi (Zhang et al., 2014). Coolen et al. (2016) investigated Arabidopsis transcriptome under infection of the necrotrophic fungus, Botrytis cinerea, alone or in combination with drought. DEGs responding to chitin, defence response, incompatible interactions, and RNA methylation were upregulated, and photosynthetic ones were deregulated up to 1 day after infection. Massive upregulation of core DEGs coding proteins that respond to oxygen-containing compounds according to the impact of drought. Kumar et al. (2022) compared two transcriptomic data sets of maize (Zea mays) silk infected with different fungal species. Set A contained data for silk samples affected by F. graminearum and U. maydis, while set B contained data from samples infected with F. verticillioides and Trichoderma atroviride. Interestingly, only 21 DEGs were found in all variants tested. Among these DEGs, peroxidase genes that control the lengthening of the germ tube to protect maize kernels from fungal disease were upregulated. The differential expression pattern was also found for the osmotine-like protein gene, which improves host defense and immune defense against stress, and for genes coding the receptor-like protein kinase subfamily. These genes appeared to be upregulated under infection with Fusarium verticillioides, Fusarium graminearum, and Ustilago maydis and downregulated when samples infected with T. atroviride.
Generally, host DEGs specifically regulated during fungal infections code proteins related to α-linolenic acid metabolism, aminoacid biosynthesis, cell wall organisation, chitin response, osmotin-like protein activity, pollen-pistil interactions, receptor-like protein kinases, resistance genes, RNA methylation, sterol biosynthesis process, as well as valine, leucine, and isoleucine degradation. SA signaling is believed to be mainly involved in resistance to biotrophic and hemibiotrophic pathogens. On the contrary, JA and ethylene signaling appeared to be indispensable for necrotrophic resistance (Pieterse et al., 2009; Guo et al., 2018). In addition, ERF belong to main TFs specifically regulating gene expression of plant host under fungal infections (Figure 3; Zhang et al., 2014; Coolen et al., 2016; Guo et al., 2018; Kumar et al., 2022).
Bacterial infections
Analysis of rice (Oryza indica) infected with Xanthomonas oryzae revealed multiple upregulated genes that encode proteins involved in signal transduction, carbohydrate metabolism, and transcription regulation. On the contrary, the downregulated genes encoded TFs and proteins necessary for lipid catabolism, oxidative burst, and cell cycle (Kottapalli et al., 2007). Furthermore, analysis of the transcriptome of tomato (Solanum lycopersicon) infected with Clavibacter michiganensis allowed the identification of upregulated genes that encode proteins that also participated in hormonal signaling, but also in protein phosphorylation, and the plant defence response (including numerous TFs, such as WRKY, NAC, HSF, and CBP60). Resistance gene analogues (RGA) that included RLK genes were also upregulated. Exogenous treatment with SA resulted in induction of genes for WRKY TFs, therefore, SA-driven gene expression resulted in improved quality of the plant immune response (Yokotani et al., 2021).
Analysis of transcriptomes of rice (O. sativa) cultivars resistant and susceptible to infection with Xanthomonas oryzaena, which causes a cereal disease called bacterial leaf streak (BLS), revealed more DEGs among the infection-susceptible cultivar. Upregulations of genes encoding proteins involved in secondary metabolism, as well as participation of selected WRKY, NAC, MYB, and bHLH TFs in plant response to bacterial infection were notable (Lu et al., 2020). Deng et al. (2023) study on resistant (“IBL2353”) and susceptible (“Ohio88119”) tomato (S. lycopersicum) cultivars infected by C. michiganensis identified new gene families that participate in antibacterial defence. The key role of WRKY TFs in this process was revealed. At least 25 genes for proteins associated with the plant defense response and WAKL20 gene (for the wall-associated receptor kinase similar to wall 20 and the only member of the WAKS subfamily that participates in innate resistance to pathogens) were upregulated in “IBL2353” cultivar. Viral-induced silencing of WAKS20 gene in the “IBL2353” cultivar resulted in the appearance of susceptibility to C. michiganensis infection, suggesting an important role for the WAKS20 gene in antibacterial defence (Deng et al., 2023).
In general, plant host genes specifically affected by bacterial infections code proteins related to the cell cycle, lipid catabolism, oxidoreductase activity, and positive regulation of ubiquitin protein ligase activity as well as transferase activity. Hormone signaling is also important for the antibacterial response of host plants (Kottapalli et al., 2007; Yokotani et al., 2021). Additionally, the host response to bacterial infections specifically mobilizes MYB and NAC TFs (Figure 3; Kottapalli et al., 2007; Lu et al., 2020; Yokotani et al., 2021; Deng et al., 2023).
Viral and viroid infections
Plant transcriptomes also respond to the plethora of viral and viroids, which may leave their genome fragments within the host genome/transcriptome (Jo et al., 2018; Mifsud et al., 2022; Raza and Wu, 2022). The enriched functional terms for genes affected by viral and viroid infections are presented in Figure 3 and the summary of quantitative data from discussed experimental reports is available in Supplementary Table S1. Most viral proteins interact with host proteins, which promote the appearance of symptoms. Plants counteract this initial symptom development by using adaptive immunity; defence hormones are also active, resulting in hampering of virus biogenesis (Malavika et al., 2023).
Viroids are among the smallest and most infectious pathogens of crops, having short ssRNA genomes. They can affect host genes whose products participate in defense response, phytohormone signaling, cell wall modification, photosynthesis, secondary metabolism, transport, gene expression, and protein modification (Joubert et al., 2022).
Tobacco transcriptomes (Nicotiana tabacum) infected with seven genotypes of tobacco etch potyvirus (TEV) varying in fitness were compared. Host genes (including those for hormone signaling and RNA silencing-mediated pathways of plant defense) whose expression was correlated with the fitness level of TEV were examined (Cervera et al., 2018). The relevance of hormone signalling and signal transduction in the host response under virus attack was also pinpointed by other reports. Recently, Li Z. et al. (2023) focused on the analysis of antiviral response of bottle gourd (Lagenaria siceraria) under cucumber green mottle mosaic virus (CGMMV) infection. Affected DEGs were involved in the biosynthesis of secondary metabolites, hormone signal transduction (JA biogenesis), plant–pathogen interactions, and carbohydrate metabolism. Méndez-López et al. (2023) investigated the impact of the pandemic infection of the Pepino mosaic virus (PepMV) on two tomato cultivars. The SlGSTU38 protein belongs to PepMV-specific susceptibility factors. The transcriptomes of healthy and virus-infected knocked out plants (gstu38) were examined. When gstu38 plants were compared with healthy wild-type plants, some key stress-related genes (including those for WRKY TFs) were upregulated and genes for intracellular signal transduction proteins, various TFs, HSP70, and proteins involved in sugar metabolism and transport were downregulated. Among DEGs affected in both tomato cultivars, genes for peroxidases, various kinases, RNA binding proteins, resistance proteins, PDH, DNA repair, recombination proteins, and chaperonins and GATA TFs were also notable. Furthermore, the rice transcriptome was assayed during planta overexpression of the OsNF-YA protein displaying antiviral defense against rice stripe virus (RSV) and Southern rice blackstreaked dwarf virus (SRBSDV). Interestingly, the expression pattern of genes for JA biogenesis was decreased in plants overexpressing NF-YA under viral infection, due to interference between the NF-YA protein and TFs that regulate JA signaling (Tan et al., 2022). On the other hand, Arabidopsis cabbage leaf curl virus (CaLCuV) infection triggered the SA-dependent pathogen response and induced the expression of genes involved in PCD, genotoxic stress, DNA repair, and cell cycle (Ascencio-Ibáñez et al., 2008).Signal transduction is also crucial when plant viruses could alter host plant traits so that they modify their insect behavior. When Myzus persicae aphids were foraging in Arabidopsis and Camelina sativa plants, cauliflower mosaic virus (CaMV) infection appeared more severe than turnip yellows virus (TuYV) and affected DEGs that encode proteins for photosynthesis, oxidation reduction proteins, microtubule-based movement, as well as enzymes for JA, ethylene, and glucosinolate biogenesis. TuYV infection in Arabidopsis plants resulted in alterations in DEGs for carbohydrate transport, defence and stress response proteins (Chesnais et al., 2022).
Zhu et al. (2018) investigated the impact of cucumber mosaic virus (CMV) infection on hot pepper (Capsicum annuum) transcriptome. Affected genes were involved in stress and defence response, and plant-pathogen interactions. DEGs for chitinase, pathogenesis-related protein (PR), tobacco mosaic virus resistance protein (TMV), WRKY TFs, and jasmonate ZIM-domain protein, were upregulated after inoculation. Intron retention for WRKY23 transcripts indicated a deep reprogramming in alternative splicing pattern under viral infection.
Zhou et al. (2019) studied transcriptomes of tomato (S. lycopersicum) plants grown from neutron-irradiated seeds and infected with tomato yellow leaf curl virus (TYLCV). Transcriptomes of plants grown from presoaked seeds were highly altered compared to those developed from dry seeds. Various doses of neutron irradiation affected the expression pattern of various DEGs. At least regulated DEGs that were common for all irradiated mutants encoded proteins for metabolism, transport, binding and responses to various stimuli, photosynthesis, and transcription. Spanò et al. (2020) applied the RNA sequence to study the transcriptomic profiles of tomato cultivars varying with resistance level, as well as their graft combinations, exposed to potato virus Y recombinant strain. Graft wounding and virus Y infection had various impacts on the tomato transcriptome, depending on genotype.
Roy et al. (2023) have used 3′RNA-seq to identify transcriptomic alterations in N. benthamiana plants infected by two strains of grapevine fanleaf virus (GFLV). During appearance of peak vein clearing symptom at 7 days post-inoculation (dpi), the genes involved in the immune response, gene regulation, and secondary metabolite production were over-represented. In other stages, DEGs related to chitinase activity, hypersensitive response, and transcriptional regulation were notable. Previously, Medzihradszky et al. (2019) investigated the transcriptomic reprogramming in the shoot apical meristem of Nicotiana benthamiana plants infected with Cymbidium ringspot virus (CymRSV). Upregulated genes coded proteins indispensable for cell defense, and downregulated DEGs were related to DNA replication and organization, shoot meristem development and plasmodesmata functions.
On the whole, plant host genes specifically affected under virus and viroid infections are related with carbohydrate transport, cell cycle regulation, DNA binding, DNA repair and recombination, DNA replication, glutathione binding, lipid metabolism, microtubule-based movement, oxidation-reduction proteins, OXPHOS activity, programmed cell death regulation, protein domain specific binding, protein folding and processing, regulation of hydrogen peroxide metabolism, senescence as well as signal transduction (with hormone signaling, highlighting relevance of JA signaling in host antiviral defence, e.g., Chesnais et al., 2022; Tan et al., 2022; Li Z. et al., 2023). Among TFs specifically mobilized under virus/viroid infection, GATA proteins were discernible (Figure 3; Ascencio-Ibáñez et al., 2008; Cervera et al., 2018; Zhu et al., 2018; Medzihradszky et al., 2019; Zhou et al., 2019; Spanò et al., 2020; Chesnais et al., 2022; Tan et al., 2022; Li Z. et al., 2023; Méndez-López et al., 2023; Roy et al., 2023).
Discussion
Transcriptomic datasets, together with proteomic and metabolomic analyses, belong to the valuable and prospective components of modern systems biology in the functional network study (Figure 1; Cramer et al., 2011). In plant cells, in addition to the transcriptome being a product of nuclear genome expression, plastid and mitochondrial transcriptomes are also present (Barkan and Goldschmidt-Clermont, 2000; Daniell et al., 2016; Rurek, 2016; Rurek et al., 2018; Best et al., 2020a). They are dynamically shaped by multiple factors at multiple levels of biological diversity (Zhu, 2002; Wang et al., 2020). Studies on plant transcriptomes, which participate in stress response, may thus allow for better understanding how plants properly react to changing environmental conditions (Zhu, 2002).
Plant stress studies employing RNA-seq
Extensive research on the plant transcriptome is being developed at various levels. Transcriptomic data can be acquired both from differential expression studies in tissue/organs or even from single cells, and modern RNA-seq approaches. From 2010 the number of experimental reports on RNA-seq and stress response among diverse plant species increased almost exponentially. In total, between 2010 and 2024 at least 4,710 plant studies related to RNA-seq and UV, drought, cold, heat and pathogen response were published (Figure 4; the data valid to 25 September 2024). This indicates the growing interest of these topics in plant molecular biology. Interestingly, reports on pathogen attack and drought using RNA-seq dominate in number from at least 2014 and those for temperature treatments (cold, heat) are generally in the third place in each year indicated (Figure 4).
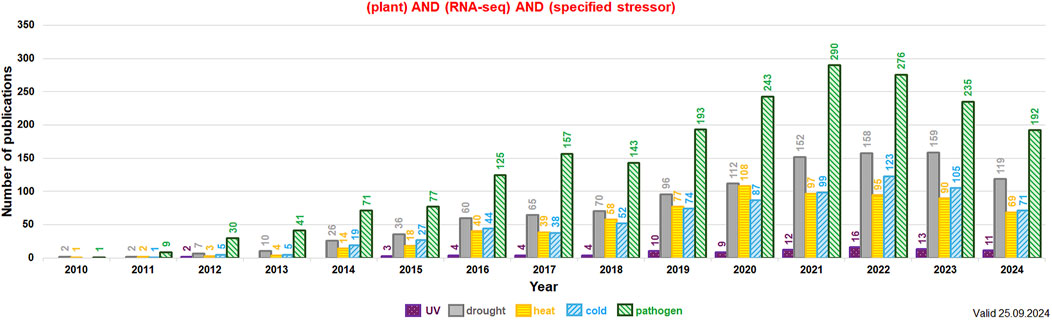
Figure 4. The detailed number of publications per year related to RNA-seq and specified stressors (values indicated above each bar; subterms indicated by the various color and checking pattern in the histogram). The key words used in the NCBI PubMed search (https://pubmed.ncbi.nlm.nih.gov/) included: plant, RNA-seq, and the given subterm (indicated in the legend below the histogram). The data for “UV” (in dark magenta), “drought” (in gray), “heat” (in orange), “cold” (in blue) and “pathogen” (in green; the joint data for bacterial, fungal and viral/viroid infections) were presented from 2010 onwards. The analysis was performed in September 2024.
Similarities and differences in transcriptomic response under various stressor conditions
The plant transcriptomic response depends greatly on the quality of the stressor and its duration, leading to metabolic flexibility (Supplementary Table S1; Wang et al., 2020; Xu et al., 2021; De Oliveira-Busatto et al., 2022). Commonly regulated genes under the action of almost all abiotic stressors often encode proteins for the photosynthetic apparatus and enzymes for secondary metabolism; interestingly, limited TFs (MYB mostly) overlap for all abiotic stressors (Figure 2).
In contrast, all conditions of biotic stress employ a large number of DEGs that represent a particularly broad landscape of enriched functional terms. DEGs for carbohydrate metabolism, defence response, protein modifications, ribosome biogenesis, transcription regulation, and secondary metabolism overlap both for bacterial, virus/viroid infections and for fungal infections across a plethora of host species (Figure 3). Furthermore, the WRKY proteins belong to universal TFs that regulate gene expression in biotic stress.
In general, photosynthesis and secondary metabolite biosynthesis belong to commonly enriched functional categories for all stress treatments discussed here.
From abiotic stressors, drought and heat stress employ the highest number of specific terms for these stressors (twelve and eleven terms, respectively, Figure 2). Drought stress appeared to be particularly detrimental at the transcriptomic level (Kumar et al., 2019; Zhu et al., 2019; Tyagi et al., 2023). Chemical treatments and UV as well as cold/freeze stress resulted in a lower quantity of terms specific to DEGs affected under action of those stressors. Furthermore, double stress treatments led to highly specific responses, sometimes increasing the number of DEGs (Zhou et al., 2019; Sewelam et al., 2020; Martin et al., 2021). It should be underlined that highly specific TFs from different families regulate gene expression under each abiotic stress conditions (with the highest number of TFs participating in drought and cold and freeze). Therefore, the most harmful stressors employ particularly numerous TFs within adaptive responses (Figure 2; Mahalingam et al., 2022).
Regarding biotic stress, virus and viral infections employ DEGs for host proteins with broadening functional categories that are specific only to those stressors. Less quantities of functional GO terms were also observed from the data representing fungal and bacterial infections (comparing with virus infections) were also notable. Genes for hormone signaling appeared to be important for both bacterial and fungal infections, and DEGs for signal transduction proteins were notably over-represented in virus/viroid infections (Figure 3). Overall, such a pattern differs from the one for abiotic stress treatments, where only a limited number of common GO terms for all treatments was evident (Figure 2). Furthermore, distinct TFs specifically regulate host gene expression under various biotic stressors, with MYB/NAC proteins mainly for bacterial infections and GATA and ERF factors for virus/virus and fungal infections, respectively (Figure 3).
As shown in Supplementary Table S1, stress-sensitive cultivars engage more DEGs in their stress responses; however, these patterns are highly tissue dependent (Mahalingam et al., 2022). Plant responses to biotic stress indicated the challenging importance of plant-microorganism interactions, which can generally influence stress tolerance (Rivero et al., 2022).
Future outlines
From the data discussed within the present review, few conclusions for future actions can be presented. Hopefully, they will increase both the quantity and quality of the data. First, more organellar transcriptomes for important crop species should be analysed to better understand the responses of the plastome and mitogenome to stress and their relevance in plant developmental steps. Furthermore, due to the peculiar under-representation of transcriptomic data on stress in some plant organs, future studies should improve such biases (Best et al., 2020a; Best et al., 2020b; Berkowitz et al., 2021).
Most of the transcriptomic data discussed here came from plant material grown under controlled conditions. Therefore, field experiments that test the variations of multiple stress responses are still awaited. Additionally, both the complexity of plant viromes and impact of the plant microbiome on stress response at the transcriptional level should be widely investigated (Jo et al., 2018; Mifsud et al., 2022; Raza and Wu, 2022).
Due to the importance of DEGs for secondary metabolite synthesis in the analysed data, it would also be important (1) to investigate further the biogenesis of active compounds, (2) to better understand how metabolic pathways contribute to various stress acclimation strategies, and (3) to develop new more stress-resistant cultivars after genetic and metabolic engineering of medicinal and crop plant species. These actions should include modern methodological tools, for example, genetic engineering and gene editing, as well as metabolome engineering (Singh et al., 2016; Yan et al., 2020; Guo et al., 2021; Kumar et al., 2023; Dong, 2024).
Author contributions
MR and MS contributed to the conception and design of the paper. MR conceived the idea for the review, supervised submission, reviewed and edited first manuscript draft. MR and MS compiled the literature and wrote first manuscript draft. MR prepared all figures and the Supplementary Material. All authors contributed to the article and approved the submitted version.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Supported by the Excellent Initiative – Research University (ID-UB) program at the Adam Mickiewicz University, Poznań and KNOW RNA Research Center at Adam Mickiewicz University, Poznań, grant no. 01/KNOW2/2014.
Acknowledgments
We would like to thank colleagues from our Institute for the valuable remarks during the preparation of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontierspartnerships.org/articles/10.3389/abp.2024.13585/full#supplementary-material
References
Ahmad, M. (2022). Genomics and transcriptomics to protect rice (Oryza sativa. L.) from abiotic stressors: -pathways to achieving zero hunger. Front. Plant Sci. 13, 1002596. doi:10.3389/fpls.2022.1002596
Alkan, C., Sajjadian, S., and Eichler, E. E. (2011). Limitations of next-generation genome sequence assembly. Nat. Methods 8, 61–65. doi:10.1038/nmeth.1527
Ascencio-Ibáñez, J. T., Sozzani, R., Lee, T.-J., Chu, T.-M., Wolfinger, R. D., Cella, R., et al. (2008). Global analysis of Arabidopsis gene expression uncovers a complex array of changes impacting pathogen response and cell cycle during geminivirus infection. Plant Physiol. 148, 436–454. doi:10.1104/pp.108.121038
Asseng, S., Foster, I., and Turner, N. C. (2011). The impact of temperature variability on wheat yields. Glob. Change Biol. 17, 997–1012. doi:10.1111/j.1365-2486.2010.02262.x
Athanasopoulou, K., Boti, M. A., Adamopoulos, P. G., Skourou, P. C., and Scorilas, A. (2021). Third-generation sequencing: the spearhead towards the radical transformation of modern genomics. Life 12, 30. doi:10.3390/life12010030
Barkan, A., and Goldschmidt-Clermont, M. (2000). Participation of nuclear genes in chloroplast gene expression. Biochimie 82, 559–572. doi:10.1016/S0300-9084(00)00602-7
Benny, J., Pisciotta, A., Caruso, T., and Martinelli, F. (2019). Identification of key genes and its chromosome regions linked to drought responses in leaves across different crops through meta-analysis of RNA-Seq data. BMC Plant Biol. 19, 194. doi:10.1186/s12870-019-1794-y
Berkowitz, O., Xu, Y., Liew, L. C., Wang, Y., Zhu, Y., Hurgobin, B., et al. (2021). RNA-seq analysis of laser microdissected Arabidopsis thaliana leaf epidermis, mesophyll and vasculature defines tissue-specific transcriptional responses to multiple stress treatments. Plant J. 107, 938–955. doi:10.1111/tpj.15314
Best, C., Mizrahi, R., and Ostersetzer-Biran, O. (2020a). Why so complex? The intricacy of genome structure and gene expression, associated with angiosperm mitochondria, may relate to the regulation of embryo quiescence or dormancy—intrinsic blocks to early plant life. Plants 9, 598. doi:10.3390/plants9050598
Best, C., Sultan, L. D., Murik, O., and Ostersetzer, O. (2020b). Insights into the mitochondrial transcriptome landscapes of two Brassicales plant species, Arabidopsis thaliana (var. Col-0) and Brassica oleracea (var. botrytis). Endocyt. Cell Res. 30, 16–38.
Bhat, K. A., Mahajan, R., Pakhtoon, M. M., Urwat, U., Bashir, Z., Shah, A. A., et al. (2022). Low temperature stress tolerance: an insight into the omics approaches for legume crops. Front. Plant Sci. 13, 888710. doi:10.3389/fpls.2022.888710
Bita, C. E., and Gerats, T. (2013). Plant tolerance to high temperature in a changing environment: Scientific fundamentals and production of heat stress-tolerant crops. Front. Plant Sci. 4, 273. doi:10.3389/fpls.2013.00273
Booth, M. W., Breed, M. F., Kendrick, G. A., Bayer, P. E., Severn-Ellis, A. A., and Sinclair, E. A. (2022). Tissue-specific transcriptome profiles identify functional differences key to understanding whole plant response to life in variable salinity. Biol. Open 11, bio059147. doi:10.1242/bio.059147
Calixto, C. P. G., Guo, W., James, A. B., Tzioutziou, N. A., Entizne, J. C., Panter, P. E., et al. (2018). Rapid and dynamic alternative splicing impacts the Arabidopsis cold response transcriptome. Plant Cell 30, 1424–1444. doi:10.1105/tpc.18.00177
Cervantes-Pérez, S. A., Thibivillliers, S., Tennant, S., and Libault, M. (2022). Review: challenges and perspectives in applying single nuclei RNA-seq technology in plant biology. Plant Sci. 325, 111486. doi:10.1016/j.plantsci.2022.111486
Cervera, H., Ambrós, S., Bernet, G. P., Rodrigo, G., and Elena, S. F. (2018). Viral fitness correlates with the magnitude and direction of the perturbation induced in the host’s transcriptome: the tobacco etch potyvirus—tobacco case study. Mol. Biol. Evol. 35, 1599–1615. doi:10.1093/molbev/msy038
Chen, C., Ge, Y., and Lu, L. (2023). Opportunities and challenges in the application of single-cell and spatial transcriptomics in plants. Front. Plant Sci. 14, 1185377. doi:10.3389/fpls.2023.1185377
Chen, S., and Li, H. (2017). Heat stress regulates the expression of genes at transcriptional and post-transcriptional levels, revealed by RNA-seq in Brachypodium distachyon. Front. Plant Sci. 7, 2067. doi:10.3389/fpls.2016.02067
Cheng, Z., Lei, N., Li, S., Liao, W., Shen, J., and Peng, M. (2019). The regulatory effects of MeTCP4 on cold stress tolerance in Arabidopsis thaliana: A transcriptome analysis. Plant Physiol. Biochem. 138, 9–16. doi:10.1016/j.plaphy.2019.02.015
Chesnais, Q., Golyaev, V., Velt, A., Rustenholz, C., Brault, V., Pooggin, M. M., et al. (2022). Comparative plant transcriptome profiling of Arabidopsis thaliana Col-0 and Camelina sativa var. celine infested with Myzus persicae aphids acquiring circulative and noncirculative viruses reveals virus- and plant-specific alterations relevant to aphid feeding behavior and transmission. Microbiol. Spectr. 10, e0013622. doi:10.1128/spectrum.00136-22
Chinnusamy, V., Zhu, J., and Zhu, J.-K. (2007). Cold stress regulation of gene expression in plants. Trends Plant Sci. 12, 444–451. doi:10.1016/j.tplants.2007.07.002
Clauw, P., Coppens, F., De Beuf, K., Dhondt, S., Van Daele, T., Maleux, K., et al. (2015). Leaf responses to mild drought stress in natural variants of Arabidopsis. Plant Physiol. 167, 800–816. doi:10.1104/pp.114.254284
Coolen, S., Proietti, S., Hickman, R., Davila Olivas, N. H., Huang, P., Van Verk, M. C., et al. (2016). Transcriptome dynamics of Arabidopsis during sequential biotic and abiotic stresses. Plant J. 86, 249–267. doi:10.1111/tpj.13167
Cramer, G. R., Urano, K., Delrot, S., Pezzotti, M., and Shinozaki, K. (2011). Effects of abiotic stress on plants: a systems biology perspective. BMC Plant Biol. 11, 163. doi:10.1186/1471-2229-11-163
Daniell, H., Lin, C.-S., Yu, M., and Chang, W.-J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 134. doi:10.1186/s13059-016-1004-2
Deng, S., Li, Z., Liu, X., Yang, W., and Wang, Y. (2023). Comparative transcriptome analysis reveals potential genes conferring resistance or susceptibility to bacterial canker in tomato. Horticulturae 9, 242. doi:10.3390/horticulturae9020242
De Oliveira-Busatto, L. A., De Almeida, R. M. C., Weber, R. L. M., Favero, D., Bredemeier, C., Da Silva Giordano, C. P., et al. (2022). The soybean Transcriptogram allows a wide genome-to-single-gene analysis that evinces time-dependent drought response. Plant Mol. Biol. Rep. 40, 1–27. doi:10.1007/s11105-021-01297-4
Dong, H. (2024). Application of genome editing techniques to regulate gene expression in crops. BMC Plant Biol. 24, 100. doi:10.1186/s12870-024-04786-2
Dong, Y., Gupta, S., Wargent, J. J., Putterill, J., Macknight, R. C., Gechev, T. S., et al. (2023). Comparative transcriptomics of multi-stress responses in Pachycladon cheesemanii and Arabidopsis thaliana. Int. J. Mol. Sci. 24, 11323. doi:10.3390/ijms241411323
Du, J., Li, X., Li, T., Yu, D., and Han, B. (2017). Genome-wide transcriptome profiling provides overwintering mechanism of Agropyron mongolicum. BMC Plant Biol. 17, 138. doi:10.1186/s12870-017-1086-3
Evans, T. G. (2015). Considerations for the use of transcriptomics in identifying the ‘genes that matter’ for environmental adaptation. J. Exp. Biol. 218, 1925–1935. doi:10.1242/jeb.114306
Fang, S., Zhao, P., Tan, Z., Peng, Y., Xu, L., Jin, Y., et al. (2022). Combining physio-biochemical characterization and transcriptome analysis reveal the responses to varying degrees of drought stress in Brassica napus L. Int. J. Mol. Sci. 23, 8555. doi:10.3390/ijms23158555
Forster, B. P., Heberle-Bors, E., Kasha, K. J., and Touraev, A. (2007). The resurgence of haploids in higher plants. Trends Plant Sci. 12, 368–375. doi:10.1016/j.tplants.2007.06.007
Frank, G., Pressman, E., Ophir, R., Althan, L., Shaked, R., Freedman, M., et al. (2009). Transcriptional profiling of maturing tomato (Solanum lycopersicum L.) microspores reveals the involvement of heat shock proteins, ROS scavengers, hormones, and sugars in the heat stress response. J. Exp. Bot. 60, 3891–3908. doi:10.1093/jxb/erp234
Guan, Q., Lu, X., Zeng, H., Zhang, Y., and Zhu, J. (2013). Heat stress induction of miR398 triggers a regulatory loop that is critical for thermotolerance in Arabidopsis. Plant J. 74, 840–851. doi:10.1111/tpj.12169
Gull, A., Ahmad Lone, A., and Ul Islam Wani, N. (2019). “Biotic and abiotic stresses in plants,” in Abiotic and biotic stress in plants. Editor A. Bosco de Oliveira (London, United Kingdom: IntechOpen Limited). doi:10.5772/intechopen.85832
Guo, J., Huang, Z., Sun, J., Cui, X., and Liu, Y. (2021). Research progress and future development trends in medicinal plant transcriptomics. Front. Plant Sci. 12, 691838. doi:10.3389/fpls.2021.691838
Guo, W.-L., Chen, B.-H., Chen, X.-J., Guo, Y.-Y., Yang, H.-L., Li, X.-Z., et al. (2018). Transcriptome profiling of pumpkin (Cucurbita moschata Duch.) leaves infected with powdery mildew. PLoS ONE 13, e0190175. doi:10.1371/journal.pone.0190175
Hasan, M., and Bae, H. (2017). An overview of stress-induced resveratrol synthesis in grapes: perspectives for resveratrol-enriched grape products. Molecules 22, 294. doi:10.3390/molecules22020294
Hu, J., Cai, J., Xu, T., and Kang, H. (2022). Epitranscriptomic mRNA modifications governing plant stress responses: Underlying mechanism and potential application. Plant Biotechnol. J. 20, 2245–2257. doi:10.1111/pbi.13913
Huang, F., Lei, Y., Duan, J., Kang, Y., Luo, Y., Ding, D., et al. (2024). Investigation of heat stress responses and adaptation mechanisms by integrative metabolome and transcriptome analysis in tea plants (Camellia sinensis). Sci. Rep. 14, 10023. doi:10.1038/s41598-024-60411-0
Huang, N., Nie, F., Ni, P., Luo, F., Gao, X., and Wang, J. (2021). NeuralPolish: A novel Nanopore polishing method based on alignment matrix construction and orthogonal Bi-GRU networks. Bioinformatics 37, 3120–3127. doi:10.1093/bioinformatics/btab354
Imadi, S. R., Kazi, A. G., Ahanger, M. A., Gucel, S., and Ahmad, P. (2015). Plant transcriptomics and responses to environmental stress: an overview. J. Genet. 94, 525–537. doi:10.1007/s12041-015-0545-6
Jin, X., Wang, Z., Li, X., Ai, Q., Wong, D. C. J., Zhang, F., et al. (2024). Current perspectives of lncRNAs in abiotic and biotic stress tolerance in plants. Front. Plant Sci. 14, 1334620. doi:10.3389/fpls.2023.1334620
Jo, Y., Lian, S., Chu, H., Cho, J. K., Yoo, S.-H., Choi, H., et al. (2018). Peach RNA viromes in six different peach cultivars. Sci. Rep. 8, 1844. doi:10.1038/s41598-018-20256-w
Joubert, M., Van Den Berg, N., Theron, J., and Swart, V. (2022). Transcriptomics advancement in the complex response of plants to viroid infection. Int. J. Mol. Sci. 23, 7677. doi:10.3390/ijms23147677
Jung, J. H., Kim, H.-Y., Kim, H. S., and Jung, S. H. (2020). Transcriptome analysis of Panax ginseng response to high light stress. J. Ginseng Res. 44, 312–320. doi:10.1016/j.jgr.2018.12.009
Ke, J., Li, M.-T., Xu, S., Ma, J., Liu, M.-Y., and Han, Y. (2023). Advances for pharmacological activities of Polygonum cuspidatum - A review. Pharm. Biol. 61, 177–188. doi:10.1080/13880209.2022.2158349
Kenrick, P., and Crane, P. R. (1997). The origin and early diversification of land plants: a cladistic study. Washington, DC: Smithsonian Institution Press.
Kottapalli, K. R., Rakwal, R., Satoh, K., Shibato, J., Kottapalli, P., Iwahashi, H., et al. (2007). Transcriptional profiling of indica rice cultivar IET8585 (Ajaya) infected with bacterial leaf blight pathogen Xanthomonas oryzae pv oryzae. Plant Physiol. Biochem. 45, 834–850. doi:10.1016/j.plaphy.2007.07.013
Kourani, M., Mohareb, F., Rezwan, F. I., Anastasiadi, M., and Hammond, J. P. (2022). Genetic and physiological responses to heat stress in Brassica napus. Front. Plant Sci. 13, 832147. doi:10.3389/fpls.2022.832147
Kumar, A., Kanak, K. R., Arunachalam, A., Dass, R. S., and Lakshmi, P. T. V. (2022). Comparative transcriptome profiling and weighted gene co-expression network analysis to identify core genes in maize (Zea mays L.) silks infected by multiple fungi. Front. Plant Sci. 13, 985396. doi:10.3389/fpls.2022.985396
Kumar, J., Gunapati, S., Kianian, S. F., and Singh, S. P. (2018). Comparative analysis of transcriptome in two wheat genotypes with contrasting levels of drought tolerance. Protoplasma 255, 1487–1504. doi:10.1007/s00709-018-1237-x
Kumar, M., Chauhan, A. S., Kumar, M., Yusuf, M. A., Sanyal, I., and Chauhan, P. S. (2019). Transcriptome sequencing of chickpea (Cicer arietinum L.) genotypes for identification of drought-responsive genes under drought stress condition. Plant Mol. Biol. Rep. 37, 186–203. doi:10.1007/s11105-019-01147-4
Kumar, M., Prusty, M. R., Pandey, M. K., Singh, P. K., Bohra, A., Guo, B., et al. (2023). Application of CRISPR/Cas9-mediated gene editing for abiotic stress management in crop plants. Front. Plant Sci. 14, 1157678. doi:10.3389/fpls.2023.1157678
Li, L., Liang, Y., Liu, Y., Sun, Z., Liu, Y., Yuan, Z., et al. (2023). Transcriptome analyses reveal photosynthesis-related genes involved in photosynthetic regulation under low temperature stress in Lavandula angustifolia Mill. Front. Plant Sci. 14, 1268666. doi:10.3389/fpls.2023.1268666
Li, X., Liu, L., Sun, S., Li, Y., Jia, L., Ye, S., et al. (2022). Leaf-transcriptome profiles of phoebe bournei provide insights into temporal drought stress responses. Front. Plant Sci. 13, 1010314. doi:10.3389/fpls.2022.1010314
Li, Y. M., Li, S. X., Li, X. S., and Li, C. Y. (2018). Transcriptome studies with the third-generation sequencing technology. Life Sci. Instrum. 16, 114–121.
Li, Z., Tang, Y., Lan, G., Yu, L., Ding, S., She, X., et al. (2023). Transcriptome and metabolome analyses reveal that jasmonic acids may facilitate the infection of cucumber green mottle mosaic virus in bottle gourd. Int. J. Mol. Sci. 24, 16566. doi:10.3390/ijms242316566
Liu, Y., Cai, Y., Li, Y., Zhang, X., Shi, N., Zhao, J., et al. (2022). Dynamic changes in the transcriptome landscape of Arabidopsis thaliana in response to cold stress. Front. Plant Sci. 13, 983460. doi:10.3389/fpls.2022.983460
Liu, Z., Xu, J., Wu, X., Wang, Y., Lin, Y., Wu, D., et al. (2019). Molecular analysis of UV-C induced resveratrol accumulation in Polygonum cuspidatum leaves. Int. J. Mol. Sci. 20, 6185. doi:10.3390/ijms20246185
Lorence, A., Chevone, B. I., Mendes, P., and Nessler, C. L. (2004). myo -inositol oxygenase offers a possible entry point into plant ascorbate biosynthesis. Plant Physiol. 134, 1200–1205. doi:10.1104/pp.103.033936
Lowe, R., Shirley, N., Bleackley, M., Dolan, S., and Shafee, T. (2017). Transcriptomics technologies. PLoS Comput. Biol. 13, e1005457. doi:10.1371/journal.pcbi.1005457
Lu, L., Yang, D., Tang, D., Li, S., and Chen, Z. (2020). Transcriptome analysis of different rice cultivars provides novel insights into the rice response to bacterial leaf streak infection. Funct. Integr. Genomics 20, 681–693. doi:10.1007/s10142-020-00744-x
Ma, L., Coulter, J. A., Liu, L., Zhao, Y., Chang, Y., Pu, Y., et al. (2019). Transcriptome analysis reveals key cold-stress-responsive genes in winter rapeseed (Brassica rapa L.). Int. J. Mol. Sci. 20, 1071. doi:10.3390/ijms20051071
Ma, L. N., Yang, J. B., Ding, Y. F., and Li, Y. K. (2019). Research progress on three generations sequencing technology and its application. China Anim. Husb. Vet. Med. 46, 2246–2256. doi:10.16431/j.cnki.1671-7236.2019.08.007
Mahalingam, R., Duhan, N., Kaundal, R., Smertenko, A., Nazarov, T., and Bregitzer, P. (2022). Heat and drought induced transcriptomic changes in barley varieties with contrasting stress response phenotypes. Front. Plant Sci. 13, 1066421. doi:10.3389/fpls.2022.1066421
Malavika, M., Prakash, V., and Chakraborty, S. (2023). Recovery from virus infection: plant’s armory in action. Planta 257, 103. doi:10.1007/s00425-023-04137-9
Martin, R. C., Kronmiller, B. A., and Dombrowski, J. E. (2021). Transcriptome analysis of Lolium temulentum exposed to a combination of drought and heat stress. Plants 10, 2247. doi:10.3390/plants10112247
McDowell, J. M., and Dangl, J. L. (2000). Signal transduction in the plant immune response. Trends Biochem. Sci. 25, 79–82. doi:10.1016/S0968-0004(99)01532-7
Medzihradszky, A., Gyula, P., Sós-Hegedűs, A., Szittya, G., and Burgyán, J. (2019). Transcriptome reprogramming in the shoot apical meristem of CymRSV-infected Nicotiana benthamiana plants associates with viral exclusion and the lack of recovery. Mol. Plant Pathol. 20, 1748–1758. doi:10.1111/mpp.12875
Méndez-López, E., Donaire, L., Gosálvez, B., Díaz-Vivancos, P., Sánchez-Pina, M. A., Tilsner, J., et al. (2023). Tomato SlGSTU38 interacts with the PepMV coat protein and promotes viral infection. New Phytol. 238, 332–348. doi:10.1111/nph.18728
Miao, Z., Xu, W., Li, D., Hu, X., Liu, J., Zhang, R., et al. (2015). De novo transcriptome analysis of Medicago falcata reveals novel insights about the mechanisms underlying abiotic stress-responsive pathway. BMC Genomics 16, 818. doi:10.1186/s12864-015-2019-x
Mifsud, J. C. O., Gallagher, R. V., Holmes, E. C., and Geoghegan, J. L. (2022). Transcriptome mining expands knowledge of RNA viruses across the plant kingdom. J. Virol. 96, e0026022–22. doi:10.1128/jvi.00260-22
Mikołajczak, K., Kuczyńska, A., Krajewski, P., Kempa, M., and Nuc, M. (2023). Transcriptome profiling disclosed the effect of single and combined drought and heat stress on reprogramming of genes expression in barley flag leaf. Front. Plant Sci. 13, 1096685. doi:10.3389/fpls.2022.1096685
Morris, J. L., Puttick, M. N., Clark, J. W., Edwards, D., Kenrick, P., Pressel, S., et al. (2018). The timescale of early land plant evolution. Proc. Natl. Acad. Sci. U.S.A. 115, E2274-E2283. doi:10.1073/pnas.1719588115
National Center for Biotechnology Information (2024). National Center for Biotechnology Information. Available at: https://pubmed.ncbi.nlm.nih.gov/(Accessed September 25, 2024).
Naydenov, N. G., Khanam, S., Siniauskaya, M., and Nakamura, C. (2010). Profiling of mitochondrial transcriptome in germinating wheat embryos and seedlings subjected to cold, salinity and osmotic stresses. Genes Genet. Syst. 85, 31–42. doi:10.1266/ggs.85.31
Obaid, A. Y., Sabir, J. S. M., Atef, A., Liu, X., Edris, S., El-Domyati, F. M., et al. (2016). Analysis of transcriptional response to heat stress in Rhazya stricta. BMC Plant Biol. 16, 252. doi:10.1186/s12870-016-0938-6
Pieterse, C. M. J., Leon-Reyes, A., Van der Ent, S., and Van Wees, S. C. M. (2009). Networking by small-molecule hormones in plant immunity. Nat. Chem. Biol. 5, 308–316. doi:10.1038/nchembio.164
Rashid, F. A. A., Crisp, P. A., Zhang, Y., Berkowitz, O., Pogson, B. J., Day, D. A., et al. (2020). Molecular and physiological responses during thermal acclimation of leaf photosynthesis and respiration in rice. Plant, Cell & Environ. 43, 594–610. doi:10.1111/pce.13706
Raza, A., and Wu, Q. (2022). “Diagnosis of viral diseases using deep sequencing and metagenomics analyses,” in Plant virology. Editors A. Wang, and Y. Li (New York, NY: Springer US), 225–243. doi:10.1007/978-1-0716-1835-6_22
Rivero, R. M., Mittler, R., Blumwald, E., and Zandalinas, S. I. (2022). Developing climate-resilient crops: improving plant tolerance to stress combination. Plant J. 109, 373–389. doi:10.1111/tpj.15483
Roy, B. G., DeBlasio, S., Yang, Y., Thannhauser, T., Heck, M., and Fuchs, M. (2023). Profiling plant proteome and transcriptome changes during grapevine fanleaf virus infection. J. Proteome Res. 22, 1997–2017. doi:10.1021/acs.jproteome.3c00069
Rurek, M. (2016). Participation of non-coding RNAs in plant organelle biogenesis. Acta Biochim. Pol. 63, 653–663. doi:10.18388/abp.2016_1346
Rurek, M., Czołpińska, M., Pawłowski, T., Krzesiński, W., and Spiżewski, T. (2018). Cold and heat stress diversely alter both cauliflower respiration and distinct mitochondrial proteins including OXPHOS components and matrix enzymes. Int. J. Mol. Sci. 19, 877. doi:10.3390/ijms19030877
Sato, M., Tsuda, K., Wang, L., Coller, J., Watanabe, Y., Glazebrook, J., et al. (2010). Network modeling reveals prevalent negative regulatory relationships between signaling sectors in Arabidopsis immune signaling. PLoS Pathog. 6, e1001011. doi:10.1371/journal.ppat.1001011
Schliesky, S., Gowik, U., Weber, A. P. M., and Bräutigam, A. (2012). RNA-seq assembly – are we there yet? Front. Plant Sci. 3, 220. doi:10.3389/fpls.2012.00220
Sewelam, N., Brilhaus, D., Bräutigam, A., Alseekh, S., Fernie, A. R., and Maurino, V. G. (2020). Molecular plant responses to combined abiotic stresses put a spotlight on unknown and abundant genes. J. Exp. Bot. 71, 5098–5112. doi:10.1093/jxb/eraa250
Shanker, A. K., Maheswari, M., Yadav, S. K., Desai, S., Bhanu, D., Attal, N. B., et al. (2014). Drought stress responses in crops. Funct. Integr. Genomics 14, 11–22. doi:10.1007/s10142-013-0356-x
Sharma, R., Singh, G., Bhattacharya, S., and Singh, A. (2018). Comparative transcriptome meta-analysis of Arabidopsis thaliana under drought and cold stress. PLoS ONE 13, e0203266. doi:10.1371/journal.pone.0203266
Singh, S., Parihar, P., Singh, R., Singh, V. P., and Prasad, S. M. (2016). Heavy metal tolerance in plants: role of transcriptomics, proteomics, metabolomics, and ionomics. Front. Plant Sci. 6, 1143. doi:10.3389/fpls.2015.01143
Son, S., and Park, S. R. (2023). Plant translational reprogramming for stress resilience. Front. Plant Sci. 14, 1151587. doi:10.3389/fpls.2023.1151587
Song, Y., Ma, B., Guo, Q., Zhou, L., Lv, C., Liu, X., et al. (2022). UV-B induces the expression of flavonoid biosynthetic pathways in blueberry (Vaccinium corymbosum) calli. Front. Plant Sci. 13, 1079087. doi:10.3389/fpls.2022.1079087
Spanò, R., Ferrara, M., Montemurro, C., Mulè, G., Gallitelli, D., and Mascia, T. (2020). Grafting alters tomato transcriptome and enhances tolerance to an airborne virus infection. Sci. Rep. 10, 2538. doi:10.1038/s41598-020-59421-5
Sprang, M., Andrade-Navarro, M. A., and Fontaine, J.-F. (2022). Batch effect detection and correction in RNA-seq data using machine-learning-based automated assessment of quality. BMC Bioinformatics 23, 279. doi:10.1186/s12859-022-04775-y
Suzuki, N., Bassil, E., Hamilton, J. S., Inupakutika, M. A., Zandalinas, S. I., Tripathy, D., et al. (2016). ABA is required for plant acclimation to a combination of salt and heat stress. PLoS ONE 11, e0147625. doi:10.1371/journal.pone.0147625
Tan, X., Zhang, H., Yang, Z., Wei, Z., Li, Y., Chen, J., et al. (2022). NF-YA transcription factors suppress jasmonic acid-mediated antiviral defense and facilitate viral infection in rice. PLoS Pathog. 18, e1010548. doi:10.1371/journal.ppat.1010548
Tu, M., Du, C., Yu, B., Wang, G., Deng, Y., Wang, Y., et al. (2023). Current advances in the molecular regulation of abiotic stress tolerance in sorghum via transcriptomic, proteomic, and metabolomic approaches. Front. Plant Sci. 14, 1147328. doi:10.3389/fpls.2023.1147328
Tyagi, A., Kumar, S., and Mohapatra, T. (2023). Biochemical, physiological and molecular responses of rice to terminal drought stress: transcriptome profiling of leaf and root reveals the key stress-responsive genes. J. Plant Biochem. Biotechnol. doi:10.1007/s13562-023-00865-x
Tyagi, P., Singh, D., Mathur, S., Singh, A., and Ranjan, R. (2022). Upcoming progress of transcriptomics studies on plants: an overview. Front. Plant Sci. 13, 1030890. doi:10.3389/fpls.2022.1030890
Umar, O., Ranti, L., Abdulbaki, A., Bola, A., Abdulhamid, A., and Biola, M. (2021). “Stresses in plants: biotic and abiotic,” in Current trends in wheat research. Editor Mahmood-ur-Rahman Ansari (London, United Kingdom: IntechOpen Limited). Available at: www.intechopen.com/chapters/79012.
Valdés-López, O., Batek, J., Gomez-Hernandez, N., Nguyen, C. T., Isidra-Arellano, M. C., Zhang, N., et al. (2016). Soybean roots grown under heat stress show global changes in their transcriptional and proteomic profiles. Front. Plant Sci. 7, 517. doi:10.3389/fpls.2016.00517
Vashisth, D., Kumar, R., Rastogi, S., Patel, V. K., Kalra, A., Gupta, M. M., et al. (2018). Transcriptome changes induced by abiotic stresses in Artemisia annua. Sci. Rep. 8, 3423. doi:10.1038/s41598-018-21598-1
Verma, S., Nizam, S., and Verma, P. K. (2013). “Biotic and abiotic stress signaling in plants,” in: Stress signaling in plants: genomics and proteomics perspective, volume 1. Editors M. Sarwat, A. Ahmad, and M. Abdin (New York, NY: Springer New York), 1, 25–49. doi:10.1007/978-1-4614-6372-6_2
Vicente, R., Bolger, A. M., Martínez-Carrasco, R., Pérez, P., Gutiérrez, E., Usadel, B., et al. (2019). De novo transcriptome analysis of durum wheat flag leaves provides new insights into the regulatory response to elevated CO2 and high temperature. Front. Plant Sci. 10, 1605. doi:10.3389/fpls.2019.01605
Wang, H., Xu, Y., Zhang, Z., Zhang, G., Tan, C., and Ye, L. (2024). Development and application of transcriptomics technologies in plant science. Crop Des. 3, 100057. doi:10.1016/j.cropd.2024.100057
Wang, X., Li, N., Li, W., Gao, X., Cha, M., Qin, L., et al. (2020). Advances in transcriptomics in the response to stress in plants. Glob. Med. Genet. 07, 030–034. doi:10.1055/s-0040-1714414
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi:10.1038/nrg2484
Wu, L., Taohua, Z., Gui, W., Xu, L., Li, J., and Ding, Y. (2015). Five pectinase gene expressions highly responding to heat stress in rice floral organs revealed by RNA-seq analysis. Biochem. Biophysical Res. Commun. 463, 407–413. doi:10.1016/j.bbrc.2015.05.085
Xia, K., Wu, Q., Yang, Y., Liu, Q., Wang, Z., Zhao, Z., et al. (2024). Drought stress induced different response mechanisms in three Dendrobium species under different photosynthetic pathways. Int. J. Mol. Sci. 25, 2731. doi:10.3390/ijms25052731
Xing, X., Ding, Y., Jin, J., Song, A., Chen, S., Chen, F., et al. (2021). Physiological and transcripts analyses reveal the mechanism by which melatonin alleviates heat stress in Chrysanthemum seedlings. Front. Plant Sci. 12, 673236. doi:10.3389/fpls.2021.673236
Xu, W., Li, R., Zhang, N., Ma, F., Jiao, Y., and Wang, Z. (2014). Transcriptome profiling of Vitis amurensis, an extremely cold-tolerant Chinese wild Vitis species, reveals candidate genes and events that potentially connected to cold stress. Plant Mol. Biol. 86, 527–541. doi:10.1007/s11103-014-0245-2
Xu, X., Legay, S., Sergeant, K., Zorzan, S., Leclercq, C. C., Charton, S., et al. (2021). Molecular insights into plant desiccation tolerance: Transcriptomics, proteomics and targeted metabolite profiling in Craterostigma plantagineum. Plant J. 107, 377–398. doi:10.1111/tpj.15294
Yan, J., Qian, L., Zhu, W., Qiu, J., Lu, Q., Wang, X., et al. (2020). Integrated analysis of the transcriptome and metabolome of purple and green leaves of Tetrastigma hemsleyanum reveals gene expression patterns involved in anthocyanin biosynthesis. PLoS ONE 15, e0230154. doi:10.1371/journal.pone.0230154
Yang, Y., Xia, J., Fang, X., Jia, H., Wang, X., Lin, Y., et al. (2023). Drought stress in ‘Shine Muscat’ grapevine: Consequences and a novel mitigation strategy–5-aminolevulinic acid. Front. Plant Sci. 14, 1129114. doi:10.3389/fpls.2023.1129114
Yokotani, N., Hasegawa, Y., Sato, M., Hirakawa, H., Kouzai, Y., Nishizawa, Y., et al. (2021). Transcriptome analysis of Clavibacter michiganensis subsp. michiganensis-infected tomatoes: a role of salicylic acid in the host response. BMC Plant Biol. 21, 476. doi:10.1186/s12870-021-03251-8
Young, L. W., Wilen, R. W., and Bonham-Smith, P. C. (2004). High temperature stress of Brassica napus during flowering reduces micro- and megagametophyte fertility, induces fruit abortion, and disrupts seed production. J. Exp. Bot. 55, 485–495. doi:10.1093/jxb/erh038
Yu, Y., Zhang, Y., Chen, X., and Chen, Y. (2019). Plant noncoding RNAs: Hidden players in development and stress responses. Annu. Rev. Cell Dev. Biol. 35, 407–431. doi:10.1146/annurev-cellbio-100818-125218
Zhang, C., Chen, J., Huang, W., Song, X., and Niu, J. (2021). Transcriptomics and metabolomics reveal purine and phenylpropanoid metabolism response to drought stress in Dendrobium sinense, an endemic orchid species in Hainan island. Front. Genet. 12, 692702. doi:10.3389/fgene.2021.692702
Zhang, H. (2019). The review of transcriptome sequencing: principles, history and advances. IOP Conf. Ser. Earth Environ. Sci. 332, 042003. doi:10.1088/1755-1315/332/4/042003
Zhang, H., Yang, Y., Wang, C., Liu, M., Li, H., Fu, Y., et al. (2014). Large-scale transcriptome comparison reveals distinct gene activations in wheat responding to stripe rust and powdery mildew. BMC Genomics 15, 898. doi:10.1186/1471-2164-15-898
Zhang, X., Ding, X., Ji, Y., Wang, S., Chen, Y., Luo, J., et al. (2018). Measurement of metabolite variations and analysis of related gene expression in Chinese liquorice (Glycyrrhiza uralensis) plants under UV-B irradiation. Sci. Rep. 8, 6144. doi:10.1038/s41598-018-24284-4
Zhou, H., Yu, L., Liu, S., Zhu, A., Yang, Y., Chen, C., et al. (2023). Transcriptome comparison analyses in UV-B induced AsA accumulation of Lactuca sativa L. BMC Genomics 24, 61. doi:10.1186/s12864-023-09133-7
Zhou, Y., Cho, W. K., Byun, H., Kil, E., Bak, S., Moon, D., et al. (2019). Transcriptome profiles of tomato plants after neutron irradiation and infection with TYLCV. Physiol. Plant. 165, 427–441. doi:10.1111/ppl.12913
Zhu, C., Li, X., and Zheng, J. (2018). Transcriptome profiling using Illumina- and SMRT-based RNA-seq of hot pepper for in-depth understanding of genes involved in CMV infection. Gene 666, 123–133. doi:10.1016/j.gene.2018.05.004
Zhu, H., Zhou, Y., Zhai, H., He, S., Zhao, N., and Liu, Q. (2019). Transcriptome profiling reveals insights into the molecular mechanism of drought tolerance in sweetpotato. J. Integr. Agric. 18, 9–23. doi:10.1016/S2095-3119(18)61934-3
Zhu, J.-K. (2002). Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 53, 247–273. doi:10.1146/annurev.arplant.53.091401.143329
Glossary
ABA abscisic acid
ABI3VP1 ABSCISIC ACID INSENSITIVE 3 VIVIPAROUS 1
ABF ABSCISIC ACID RESPONSIVE ELEMENT-BINDING FACTOR
ACC 1-aminocyclopropane-1-carboxylic oxidase
ALA 5-aminolevulinic acid
AOS allene oxide synthase
AOX alternative oxidase
AP APETALA
APX ascorbate peroxidase
ARF auxin response factor
ARID AT-rich interactive domain
ASC L-ascorbic acid
AUX auxin receptor
BBR-BPC Barley B Recombinant/Basic PentaCysteine
BIM BES1-INTERACTING MYC-LIKE
bHLH basic helix–loop–helix
BLS bacterial leaf streak
BPM BTB/POZ and MATH domain-containing protein
BT BTB and TAZ domain protein
bHLH basic helix-loop-helix
bZIP basic (region) leucine zipper
CaLCuV cabbage leaf curl virus
CAM crassulacean acid metabolism
CaMV cauliflower mosaic virus
CAT catalase
CBP calmodulin-binding protein
CDP CAAT displacement protein (transcriptional repressor)
CGMMV cucumber green mottle mosaic virus
C2H zinc finger protein
C3H zinc finger CCCH domain-containing protein
C4H 4-cinnamate hydroxylase
CHI chalcone isomerase
CHS chalcone synthase
4CL 4-coumarate: CoA ligase
Clp caseinolytic protease
CLP chaperonin-like protein
CMV cucumber mosaic virus
CRISPR/Cas clustered regularly interspaced short palindromic repeats/CRISPR-associated
CRK cysteine-rich receptor-like kinase
CXIP CAX-interacting protein
CymRSV Cymbidium ringspot virus
DEGs differentially-expressed genes
DIMI DIMINUTO
Dof DNA-binding with one finger
EIL ethylene insensitive-like
ER endoplasmic reticulum
EREBP ethylene-responsive element binding protein
ERF ethylene response factor
ETI effector-triggered immunity
FLS flavonol synthase
FRS FAR1-RELATED SEQUENCE
FT FLOWERING LOCUS T
G2-like GOLDEN2-LIKE
GABA γ-aminobutyric acid
GARP GOLDEN2, ARR-B and Psr1 superfamily
GFLV grapevine fanleaf virus
GH3 Gretchen Hagen3
GID GA-INSENSITIVE DWARF
GRAS GIBBERELLIC-ACID INSENSITIVE, REPRESSOR of GAI and SCARECROW
GRF growth regulating factor
GSTU glutathione S-transferase belonging to the tau class
HAB HYPERSENSITIVE TO ABSCISIC ACID
HAP CCAAT-box binding protein
HD homeodomain
HLH helix-loop-helix
HR hypersensitive response
HSF heat shock factor
HSP heat shock protein
JA jasmonic acid
LEA late-embryogenesis abundant
LHC light-harvesting complex
LIM homeobox transcription factor
LOX lipoxygenase
MADS MINICHROMOSOME MAINTENANCE 1/AGAMOUS/DEFICIENS/SERUM RESPONSE FACTOR
MAPK mitogen-associated protein kinase
MDHAR monodehydroascorbate reductase
MIOX myo-inositol oxygenase
MYB myeloblastosis
NAC no apical meristem/ATAF1/cup-shaped cotyledon
NF-Y nuclear factor Y
NIN nodule inception
OXPHOS oxidative phosphorylation
PAL phenylalanine ammonia-lyase
PAMP pathogen-associated molecular pattern
PCD programmed cell death
PcG Polycomb group
PDH proline dehydrogenase
PEPC phosphoenolpyruvate carboxylase
PepMV pepino mosaic virus
PLATZ plant AT-rich sequence and zinc-binding protein
POD peroxidase
PR pathogen-related
PS photosystem
PTI PAMP-triggered immunity
RGA resistance gene analogue
RLK receptor-like kinase
ROS reactive oxidative species
RR response regulator
RSV rice stripe virus
RWP-RK transcription factor with RWPxRK motif
SA salicylic acid
SBP SQUAMOSA-PROMOTER BINDING PROTEIN
SOD superoxide dismutase
SMRT single-molecule real-time
SRBSDV Southern rice blackstreaked dwarf virus
STS stilbene synthase
TCP bHLH DNA-binding domain
TEV tobacco etch potyvirus
TF transcription factor
TMV tobacco mosaic virus
TUB Tubby family of bipartite transcription factors
TuYV turnip yellows virus
TYLCV tomato yellow leaf curl virus
UFGT UDP-glucose:flavonoid 3-O-glucosyltransferase
USP universal stress protein
WAKL wall-associated receptor kinase-like
WAKS wall-associated kinases
WRKY transcription factor family
WT wild type
WUS WUSCHEL
ZIM zinc-finger inflorescence meristem
ZIP leucine zipper
Keywords: acclimation, differentially expressed genes, plant transcriptome, RNA-seq, stress response
Citation: Rurek M and Smolibowski M (2024) Variability of plant transcriptomic responses under stress acclimation: a review from high throughput studies. Acta Biochim. Pol 71:13585. doi: 10.3389/abp.2024.13585
Received: 26 July 2024; Accepted: 15 October 2024;
Published: 25 October 2024.
Edited by:
Maurycy Daroch, Peking University, ChinaReviewed by:
Małgorzata Ryngajłło, Lodz University of Technology, PolandAgnieszka Zmienko, Polish Academy of Sciences, Poland
Copyright © 2024 Rurek and Smolibowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michał Rurek, cnVyZWtAYW11LmVkdS5wbA==