 Sora Kwon
Sora Kwon Reshani Jeyaratnam
Reshani Jeyaratnam Kyoung-Han Kim
Kyoung-Han Kim- 1University of Ottawa Heart Institute, Ottawa, ON, Canada
- 2Department of Cellular and Molecular Medicine, Faculty of Medicine, University of Ottawa, Ottawa, ON, Canada
- 3Translational and Molecular Medicine, Faculty of Medicine, University of Ottawa, Ottawa, ON, Canada
Metabolic dysfunction-associated steatotic liver disease (MASLD) is a metabolic disorder marked by excessive accumulation of lipids within the liver. If untreated, this condition can progress to metabolic dysfunction-associated steatohepatitis (MASH), fibrosis, cirrhosis, and ultimately, hepatocellular carcinoma (HCC). Given the liver’s pivotal role in glucose and fatty acid metabolism, disruptions in these processes are commonly observed in MASLD. Ketone bodies, crucial energy metabolites primarily produced in the liver, are also closely related to the progression of MASLD. Recent studies have demonstrated that disrupted ketogenesis not only accompanies MASLD, but may also play a causal role in its development and progression. Moreover, activation of the ketogenic pathway has been suggested as a promising strategy for reducing excessive hepatic fat accumulation. This review focuses on the regulation of ketogenesis in MASLD, emphasizing the significance of dietary and pharmacological interventions as potential therapeutic approaches to treat fatty liver disease.
Introduction
Metabolic dysfunction-associated steatotic liver disease (MASLD), formerly known as non-alcoholic fatty liver disease (NAFLD), is a prevalent chronic liver disease [1, 2], globally affecting human health with an estimated prevalence of 32% [3]. This condition is characterized by increased fat accumulation within the liver, compromising its function. The prolonged accumulation of hepatic fat in MASLD can lead to severe conditions, such as metabolic dysfunction-associated steatohepatitis (MASH), cirrhosis, and hepatocellular carcinoma (HCC). This progression is driven by lipotoxicity, leading to increased hepatic oxidative stress and the development of MASH [4]. Concurrently, increases in free fatty acid uptake and oxidative stress activate resident liver macrophages, which promote inflammation through various signaling pathways, including Toll-like receptor (TLR) 4-mediated production of pro-inflammatory cytokines [5, 6]. As the liver attempts to repair itself amid heightened inflammation, fibrosis emerges, characterized by the accumulation of extracellular matrix proteins, tissue scarring and immune cell infiltration [7, 8]. This persistent tissue scarring and immune activity eventually culminate into cirrhosis, marked by hepatocyte apoptosis [9] and impaired regenerative capacity [10]. Additionally, the elevated pro-inflammatory cytokine TNF has been associated with tumor promotion, as it stimulates hepatocyte proliferation, which can trigger the development of HCC [11]. As MASLD and its pathological progression arise from complex interactions of various factors affecting a broad spectrum of individuals, numerous studies have focused on elucidating the mechanisms driving the progression of this disease and developing effective therapeutic strategies.
Nevertheless, current treatment approaches for fatty liver disease, aside from lifestyle modifications such as weight management, dietary interventions, and exercise, are relatively limited. Insulin sensitizers, lipid-lowering medications, and antioxidants have been tested, but have not proven effective. Notably, drugs used for type 2 diabetes, such as metformin and sodium-glucose cotransporter-2 inhibitors (SGLT2i), have shown efficacy in treating fatty liver disease [12–14]. It remains unclear, though, whether the beneficial effects of these drugs on fatty liver disease are due to direct targeting of the liver function or are indirectly achieved through improved glucose homeostasis. Recently, the U.S. Food and Drug Administration approved resmetirom (Rezdiffra), a thyroid hormone receptor β (THRβ) agonist, as the first drug to directly target the liver for the treatment of MASH and moderate-to-advanced hepatic fibrosis. However, only 20%–30% of patients have shown improvement in key liver pathology indicators, and the long-term safety of resmetirom has not yet been assessed in clinical trials [15]. Therefore, the need to identify novel therapeutic targets for treating fatty liver disease remains a pressing and unmet challenge.
Dysregulated ketone body metabolism in fatty liver disease
Metabolic remodelling is a molecular and cellular hallmark in fatty liver diseases, which includes alterations in de novo lipogenesis, hepatic very-low-density lipoprotein secretion and lipoprotein metabolism, and gluconeogenesis [16]. Another notable change is the dysregulation of ketone body metabolism. In the early stage of fatty liver disease like simple steatosis, an increase in plasma ketone bodies is often observed as a result of the liver converting excessive fatty acids into ketone bodies to alleviate metabolic stress [17, 18]. However, as MASLD advances to more severe stages like MASH, levels of plasma ketone bodies in patients decrease [19]. This decline is attributed to impaired ketogenesis, a process of synthesizing water-soluble ketone bodies, such as β-hydroxybutyrate (BHB), acetoacetate (AcAc), and acetone, primarily in the liver, as fasting-induced ketosis is significantly reduced in humans with MASLD [20, 21]. In addition, the rate of ketogenesis, specifically the production of BHB and not AcAc, is negatively associated with the degree of hepatic triglyceride content [20]. Impaired ketogenesis in severe MASLD has also been consistently observed in both preclinical mouse models and humans [22, 23].
Ketone bodies are primarily generated in the liver during glucose-deprived conditions. Acetyl-CoA, mainly derived from fatty acids through beta-oxidation, undergoes a series of enzymatic reactions within the mitochondria. These reactions involve acetoacetyl-CoA thiolase (ACAT1), 3-hydroxy-3 methyglutaryl-CoA synthase 2 (HMGCS2) and HMG-CoA lyase (HMGCL), generating AcAc as a primary ketone body metabolite. AcAc is then further converted to BHB by β-hydroxybutyrate dehydrogenase (BDH1) [24–26]. Among these critical enzymes in the ketogenic pathways, HMGCS2 is notably implicated in dysregulated ketogenesis in fatty liver disease. In mice with high-fat diet (HFD)-induced MASLD, the fasting-induced increases in HMGCS2 transcript and protein are largely abolished [22]. Similarly, HMGCS2 expression is suppressed with more advanced steatotic stages, such as cirrhosis and HCC [27, 28].
Importantly, dysregulated ketogenesis is not simply an outcome but plays a causal role in the development of fatty liver disease. In infants, deficiencies in HMGCS2 or HMGCL lead to hepatomegaly and hepatic steatosis [29–31]. Consistently, postnatal mice lacking Hmgcs2 gene spontaneously develop fatty liver disease [22, 32]. The impaired hepatic ketogenic conduit by Hmgcs2 ablation causes excessive accumulation of acetyl-CoA [32, 33]. This, in turn, enhances de novo lipogenesis, hepatic glucose production, and acetylation of mitochondrial proteins, which collectively contribute to steatosis and metabolic dysfunctions in the liver. In addition, altered hepatic ketogenesis and ketone body metabolism contribute to the progression of fatty liver disease by modulating inflammation and fibrosis. For instance, ketogenic insufficiency induced by antisense oligonucleotide (ASO)-mediated Hmgcs2 knockdown in HFD-fed adult mice results in not only elevated hepatic triacylglycerol concentrations but also inflammation and injury with macrophage accumulation in the liver, characteristics of MASH [34–36]. Also, disturbance in hepatocyte-macrophage ketone body communication, specifically via AcAc (not BHB), leads to hepatic fibrosis by activating hepatic stellate cells [37]. Furthermore, hepatic deletion of monocarboxylate transporter 1 (MCT1, encoded by Slc16a1), one of the main transporters of ketone bodies [38], exacerbates hepatic steatosis in female mice [39], although it is unclear whether this aggravation of the fatty liver is mediated by impaired ketone body transport. Disruptions in key regulators of ketogenesis, including hormones such as insulin and glucagon and transcriptional regulators like PPARα and mTORC1 [40], also contribute to the development of fatty liver disease. For example, PPARα knockout mice, which exhibit impaired ketogenesis with decreased ketogenic enzymes, Hmgcs2 and Bdh1, develop hepatic steatosis [41–43]. Additionally, mTORC1, which suppresses Hmgcs2 expression and ketogenesis by inhibiting the transcriptional activity of PPARα [44], is frequently activated in fatty liver disease [45]. Collectively, these findings underscore the critical role of ketone body metabolism in MASLD development and progression. Investigations into key enzymes and regulators, such as HMGCS2, BDH1, PPARα, and mTORC1, highlight the intricate interplay between ketone body metabolism and fatty liver disease.
Targeting ketone body metabolism to treat fatty liver disease
Ketone bodies primarily serve as alternative energy fuels in extrahepatic tissues - such as the heart, skeletal muscle, and brain - during various developmental and physiological conditions, including neonatal development, pregnancy, starvation, and exercise. Importantly, the multifaceted roles of ketone bodies in metabolic health have been extensively studied. They mediate cellular signaling via G-protein receptors (i.e., GPR41, GPR43 and GPR109A) and epigenetic gene regulation through post-translational modifications (PTMs), including histone modifications, such as lysine acetoacetylation and β-hydroxybytyrylation [46–48]. These mechanisms collectively exert anti-inflammatory, antioxidative and antifibrotic effects [49–53].
It is noteworthy that elevations in ketogenesis and the administration of ketone bodies can provide significant benefits against the development and progression of fatty liver disease, underscoring the substantial health implications of ketone bodies (Figure 1). Specifically, activating ketogenesis through Hmgcs2 overexpression improves HFD-induced MASLD in mice and reduces lipid accumulation in HepG2 cells [22]. Concurrently, Bdh1 overexpression in the liver ameliorates hepatic fibrosis, inflammation and apoptosis in db/db mice [54]. In addition, the exogenous administration of AcAc reduces hepatic fibrosis in mice fed a fibrogenic diet [37], while BHB supplementation lessens liver injury and exerts anti-inflammatory effects through the down-regulation of the NLRP3 inflammasome [55–57]. Similarly, dietary supplementation with ketone esters decreases MASLD and inflammation, along with a reduction in the expression of profibrotic and proinflammatory genes, such as Col1a1 and Pdgfb [58, 59]. These findings emphasize the potential therapeutic avenues for addressing MASLD and its progression by targeting ketone body metabolism. There is growing interest in utilizing dietary and pharmacological interventions to enhance ketogenesis for treating hepatic steatosis and its progression, as detailed further below.
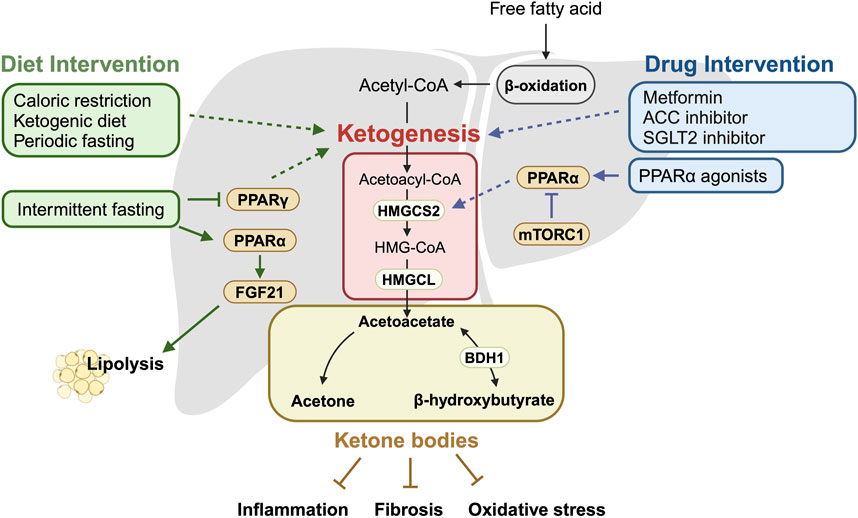
Figure 1. Summary of ketogenesis mechanisms in relation to dietary and pharmacological interventions. This schematic illustrates the key enzymes and pathways involved in ketogenesis and how they are modulated by dietary and pharmacological interventions. It highlights the impact of these interventions on the ketogenic pathway, leading to increased production of ketone bodies, which are crucial for managing MASLD. Activated ketogenesis helps reduce liver fat accumulation and inhibit fatty liver disease progression factors, such as inflammation, fibrosis, and oxidative stress. Solid arrows indicate the direction of regulatory effect, while dotted arrows represent effects that are known but not fully understood. The schematic was created with BioRender.com.
Dietary interventions
As ketogenesis has emerged as a potent target for MASLD treatment, dietary interventions that influence ketone body metabolism, such as nutritional interventions and fasting regimens, offer promising approaches for managing MASLD. Indeed, besides various positive effects on health and lifespan, nutritional interventions have demonstrated promising therapeutic impacts on MASLD with decreased hepatic triglyceride content in mice and reduced body fat and inflammation markers in humans [60, 61]. Notably, nutritional interventions, such as caloric restriction (10%–40% reduction) and ketogenic diets, effectively elevate blood ketone body levels and enhance their transport and utilization in both rodents and humans [62–65]. Specifically, caloric restriction, which entails a significant reduction in daily calorie intake, has been shown to decrease hepatic fat content [66], thereby reversing hepatic steatosis in obese rodents with metabolic diseases [60]. In MASLD patients, caloric restriction leads to reductions in fatty liver index and ALT values [67], indicating potential therapeutic benefits. Furthermore, the ketogenic diet, characterized by limited carbohydrate intake, stimulates the mobilization of fatty acids, leading to weight loss in humans and mice. It also effectively increases their blood ketone body levels while improving plasma glucose and triglycerides as well as insulin sensitivity in MASLD patients. A low-carbohydrate ketogenic diet significantly reduces intrahepatic triglyceride levels by 43.8% and alleviates hepatic inflammation and fibrosis in MASLD patients [65, 68, 69]. Consistently, ketogenic diets decrease the expression of genes involved in fatty acid synthesis while upregulating those involved in fatty acid oxidation [70–73]. These beneficial effects of ketogenic diets in the liver are mediated through hepatic fibroblast growth factor 21 (FGF21) as a regulator of the ketotic state [74, 75]. Together, these findings suggest that nutritional interventions are effective strategies for treating MASLD by promoting ketone body metabolism. However, some studies have noted that a ketogenic diet may induce hepatic steatosis, increase inflammation, and promote cellular senescence in mice [64, 76, 77]. Such discrepancies among different studies may potentially be attributed to variations in dietary composition, particularly the fat content, as well as differences in diet duration and the ages of subjects or participants. This underscores the need to carefully evaluate the potential adverse effects of ketogenic diets and understand their underlying mechanisms.
Fasting interventions, such as intermittent fasting and time-restricted feeding, which involve alternating periods of fasting and refeeding [78, 79], are effective in promoting cyclic ketogenesis, thereby potentially improving MASLD [80, 81]. Various intermittent fasting (IF) regimens, such as time-restricted feeding, alternate-day fasting, 2:1 IF, and 5:2 IF, have been shown to improve steatosis by downregulation of PPARγ, a transcription factor implicated in triglyceride homeostasis and activation of fatty acid oxidation via PPARα, in high-fat-fructose induced MASH rat models [82] and HFD-induced MASLD mice [81, 83–85]. Notably, IF also activates the hepatic autophagy-lysosome pathway, reducing hepatic lipid accumulation [84] while diminishing hepatic inflammation and fibrosis through decreased expression of IL-6 and TNFα, thereby mitigating MASH progression [81, 83, 84]. Furthermore, IF has proven effective in humans, reducing intrahepatic triglyceride content by 8.3% [86]. Collectively, these studies highlight that nutritional and fasting interventions can serve as effective therapeutic approaches for MASLD via activating ketogenesis.
Pharmacological interventions
Several pharmacological candidates have shown potential for improving fatty liver disease outcomes by affecting ketone body metabolism. These include Metformin, PPARα agonist (Fibrates), ACC (Acetyl-CoA carboxylase) inhibitors and sodium-glucose cotransporter 2 (SGLT2) inhibitors [14, 87, 88].
Metformin (1, 1-dimethylbiguanide hydrochloride) has demonstrated potential in inhibiting the progression of MASLD. Clinical studies have indicated that metformin treatment in patients with MASLD improves liver function with reductions in hepatic fat accumulation and inflammation [89, 90]. By decreasing hepatic gluconeogenesis, metformin leads to reduced blood glucose levels, which in turn suppresses the activation of lipogenic pathways and promotes hepatic ketogenesis in rat liver [91]. It has also been shown that metformin induces fasting-mimicking metabolic modification, including ketogenesis, in humans [92]. However, the specific molecular mechanism by which metformin affects hepatic ketogenesis remains unclear, and it is unknown whether the metabolic therapeutic effects of metformin are mediated through ketone bodies.
PPARα agonists, such as fibrates, play a crucial role in regulating hepatic lipid metabolism. They have been shown to upregulate the expression of genes involved in fatty liver oxidation and lipoprotein metabolism, potentially contributing to increased ketogenesis [93, 94]. By enhancing these processes, fibrates could improve liver function and reduce hepatic fat accumulation in patients with fatty liver disease. Although fenofibrate has demonstrated efficacy in improving indicators of metabolic syndrome, blood sugar levels, and hepatic function tests in clinical investigations, it has not yielded significant improvement in liver histology, including steatosis score, inflammation grade, and fibrosis stage. To address these limitations, selective PPARα modulators like Pemafibrate have been developed, which offer improved efficacy and safety profiles. Specifically, Pemafibrate has been shown to ameliorate markers of liver inflammation and fibrosis in patients with MASLD [95, 96].
Acetyl-CoA carboxylase (ACC) is a pivotal enzyme in fatty acid synthesis, catalyzing the conversion of acetyl-CoA to malonyl-CoA, a crucial step in hepatic de novo lipogenesis. Owing to its central role in lipid metabolism, ACC has emerged as a promising target for therapeutic intervention in fatty liver disease. Numerous studies have demonstrated that inhibition of ACC can effectively reduce fatty acid synthesis and, consequently, decrease hepatic lipid accumulation [97]. For example, Firsocostat (GS-0976), a liver-targeted small molecule allosteric inhibitor of ACC1/2, improves MASH in both preclinical and clinical studies [98]. Additionally, another ACC1/2 inhibitor PF-05221304, either alone or in combination with a DGAT2 (diacylglycerol O-acyltransferase 2) inhibitor, significantly reduces hepatic steatosis in patients with MASLD [99]. Furthermore, it has been shown that a small molecule IMA-1, which interrupts the arachidonate 12-lipoxygenase (ALOX12)-ACC1 interaction, decreases hepatic lipid accumulation and lowers inflammation and fibrosis in mice and macaques, addressing multiple key features of MASH [100]. Notably, a single oral dose of MK-4074, a liver-specific ACC1/2 inhibitor, increases plasma ketone bodies in mice and humans within 8 h [101], suggesting its strong ketogenic potential. Similarly, the observation that Firsocostat can increase BHB in non-hepatic cells further supports the conserved ketogenic action of ACC inhibition [102]. However, the implications of ketone bodies in ACC inhibitor-mediated hepatic protection have not been explored.
Another class of drugs that have shown promise in the context of ketogenesis and MASLD is the SGLT2 inhibitors, commonly used in the treatment of type 2 diabetes [103]. These drugs increase urinary excretion of glucose by the kidney, thereby reducing blood glucose levels. Beyond their primary use, SGLT2 inhibitors offer therapeutic benefits for MASLD by modulating key metabolic pathways. They promote lipolysis, stimulate mitochondrial biogenesis and autophagy, and reduce lipogenesis, oxidative stress, and fibrogenesis [104, 105]. Meta-analyses have also shown that SGLT2 inhibitors can reduce hepatic enzymes (e.g., ALT and AST), hepatic fat contents, and Fibrosis-4 (FIB-4) levels, suggesting they alleviate MASLD and its progression to MASH [106]. Notably, it is well known that treatment with SGLT2 inhibitors is associated with higher plasma ketone body levels in patients [104, 105]. While the exact mechanism linking SGLT2 inhibitors and ketogenesis is not fully understood [107], it has been suggested that a metabolic shift from glucose to fatty acids induced by SGLT2 inhibitors underlies ketogenesis [104]. Nevertheless, it remains unclear whether the salutary actions of SGLT2 inhibitors against MASLD are mediated by promoting ketogenesis or through SGLT2-independent actions, as observed in the failing heart [108]. Future studies are required to uncover the therapeutic mechanism of SGLT2 inhibitors for MASLD.
Additionally, Pimozide, which blocks skeletal muscle ketone oxidation, increases plasma ketone bodies and improves hyperglycemia [109], yet its effects on fatty liver disease remain unknown. Rapamycin, which inhibits mTORC1, the negative modulator of hepatic ketogenesis, also increases plasma ketone bodies [44]. However, due to its intricate actions in global metabolism and crosstalk with several pathways [110], targeting the mTOR pathway to treat fatty liver disease presents challenges. It is noteworthy that these pharmacological agents appear to promote ketogenesis indirectly, including through transcriptional activation and modulation of metabolic fluxes. The development of drug candidates that directly target ketogenic enzymes and their roles in treating fatty liver disease hold significant interest.
Discussion
In this review, we aim to summarize the current understanding of the potential role of ketogenesis as a critical player in the treatment of fatty liver disease, utilizing both dietary and drug interventions (Figure 1). The contributions of ketogenesis and ketone bodies in MASLD treatment are promising, yet further investigation is warranted to determine the extent to which the beneficial effects result from ketogenesis itself [22], the use of ketone bodies as fuel, or the cellular actions of ketone bodies as signaling molecules, or a combination of these processes [49]. In addition, careful consideration of several factors is required when evaluating treatment options that promote ketogenesis. For instance, ketoacidosis, a life-threatening complication of diabetes, has been reported as a potential side effect of both SGLT2 inhibitors [111] and ketogenic diets [112], though the underlying mechanisms are not fully understood. Furthermore, variations in the effects of ketogenic diets and intermittent fasting due to differences in sex and age have been observed [113, 114], as these factors are also known to impact ketone body metabolism [115]. Consequently, further investigation is essential to safely and effectively leverage ketone body metabolism for the treatment of fatty liver disease.
Author contributions
SK, RJ, and K-HK conceived and designed the research. All authors contributed to the article and approved the submitted version.
Funding
The authors declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the End Diabetes Award from Diabetes Canada to K-HK (OG-3-22-5697-KK). He is also a recipient of the National New Investigator Award from the Heart and Stroke Foundation of Canada (HSFC) and the Early Career Award (ER22-17-236) from the Government of Ontario, Canada. SK was supported by the MITACS Elevate Fellowship (IT34864). RJ was supported by the Natural Sciences and Engineering Research Council of Canada - Undergraduate Student Research Award (NSERC-USRA).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Eslam, M, Sanyal, AJ, George, J, Sanyal, A, Neuschwander-Tetri, B, Tiribelli, C, et al. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology (2020) 158:1999–2014.e1. doi:10.1053/j.gastro.2019.11.312
2. Rinella, ME, and Sookoian, S. From NAFLD to MASLD: updated naming and diagnosis criteria for fatty liver disease. J Lipid Res (2024) 65:100485. doi:10.1016/j.jlr.2023.100485
3. Riazi, K, Azhari, H, Charette, JH, Underwood, FE, King, JA, Afshar, EE, et al. The prevalence and incidence of NAFLD worldwide: a systematic review and meta-analysis. The Lancet Gastroenterol and Hepatol (2022) 7:851–61. doi:10.1016/S2468-1253(22)00165-0
4. Feldstein, AE, Werneburg, NW, Canbay, A, Guicciardi, ME, Bronk, SF, Rydzewski, R, et al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-α expression via a lysosomal pathway. Hepatology (2004) 40:185–94. doi:10.1002/hep.20283
5. Sharifnia, T, Antoun, J, Verriere, TGC, Suarez, G, Wattacheril, J, Wilson, KT, et al. Hepatic TLR4 signaling in obese NAFLD. Am J Physiology-Gastrointestinal Liver Physiol (2015) 309:G270–278. doi:10.1152/ajpgi.00304.2014
6. Slevin, E, Baiocchi, L, Wu, N, Ekser, B, Sato, K, Lin, E, et al. Kupffer cells: inflammation pathways and cell-cell interactions in alcohol-associated liver disease. The Am J Pathol (2020) 190:2185–93. doi:10.1016/j.ajpath.2020.08.014
7. Sanchez, JI, Parra, ER, Jiao, J, Solis Soto, LM, Ledesma, DA, Saldarriaga, OA, et al. Cellular and molecular mechanisms of liver fibrosis in patients with NAFLD. Cancers (Basel) (2023) 15:2871. doi:10.3390/cancers15112871
8. Bataller, R, and Brenner, DA. Liver fibrosis. J Clin Invest (2005) 115:209–18. doi:10.1172/JCI24282
9. Ribeiro, PS, Cortez-Pinto, H, Solá, S, Castro, RE, Ramalho, RM, Baptista, A, et al. Hepatocyte apoptosis, expression of death receptors, and activation of NF-κB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am J Gastroenterol (2004) 99:1708–17. doi:10.1111/j.1572-0241.2004.40009.x
10. Dewhurst, MR, Ow, JR, Zafer, G, van Hul, NKM, Wollmann, H, Bisteau, X, et al. Loss of hepatocyte cell division leads to liver inflammation and fibrosis. PLoS Genet (2020) 16:e1009084. doi:10.1371/journal.pgen.1009084
11. Park, EJ, Lee, JH, Yu, GY, He, G, Ali, SR, Holzer, RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell (2010) 140:197–208. doi:10.1016/j.cell.2009.12.052
12. Targher, G, Mantovani, A, and Byrne, CD. Mechanisms and possible hepatoprotective effects of glucagon-like peptide-1 receptor agonists and other incretin receptor agonists in non-alcoholic fatty liver disease. The Lancet Gastroenterol and Hepatol (2023) 8:179–91. doi:10.1016/S2468-1253(22)00338-7
13. Xing, B, Zhao, Y, Dong, B, Zhou, Y, Lv, W, and Zhao, W. Effects of sodium-glucose cotransporter 2 inhibitors on non-alcoholic fatty liver disease in patients with type 2 diabetes: a meta-analysis of randomized controlled trials. J Diabetes Invest (2020) 11:1238–47. doi:10.1111/jdi.13237
14. Farah, S, Nguyen, T, Kelsberg, G, and Safranek, S. Metformin for nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Am Fam Physician (2019) 99:262–3.
15. Harrison, SA, Bedossa, P, Guy, CD, Schattenberg, JM, Loomba, R, Taub, R, et al. A phase 3, randomized, controlled trial of resmetirom in NASH with liver fibrosis. N Engl J Med (2024) 390:497–509. doi:10.1056/NEJMoa2309000
16. Bence, KK, and Birnbaum, MJ. Metabolic drivers of non-alcoholic fatty liver disease. Mol Metab (2021) 50:101143. doi:10.1016/j.molmet.2020.101143
17. Post, A, Garcia, E, van den Berg, EH, Flores-Guerrero, JL, Gruppen, EG, Groothof, D, et al. Nonalcoholic fatty liver disease, circulating ketone bodies and all-cause mortality in a general population-based cohort. Eur J Clin Invest (2021) 51:e13627. doi:10.1111/eci.13627
18. Satapati, S, Kucejova, B, Duarte, JA, Fletcher, JA, Reynolds, L, Sunny, NE, et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J Clin Invest (2015) 125:4447–62. doi:10.1172/JCI82204
19. Mannisto, VT, Simonen, M, Hyysalo, J, Soininen, P, Kangas, AJ, Kaminska, D, et al. Ketone body production is differentially altered in steatosis and non-alcoholic steatohepatitis in obese humans. Liver Int (2015) 35:1853–61. doi:10.1111/liv.12769
20. Fletcher, JA, Deja, S, Satapati, S, Fu, X, Burgess, SC, and Browning, JD. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight (2019) 4. doi:10.1172/jci.insight.127737
21. Lee, S, Bae, J, Jo, DR, Lee, M, Lee, Y, Kang, ES, et al. Impaired ketogenesis is associated with metabolic-associated fatty liver disease in subjects with type 2 diabetes. Front Endocrinol (Lausanne) (2023) 14:1124576. doi:10.3389/fendo.2023.1124576
22. Asif, S, Kim, RY, Fatica, T, Sim, J, Zhao, X, Oh, Y, et al. Hmgcs2-mediated ketogenesis modulates high-fat diet-induced hepatosteatosis. Mol Metab (2022) 61:101494. doi:10.1016/j.molmet.2022.101494
23. Mey, JT, Erickson, ML, Axelrod, CL, King, WT, Flask, CA, McCullough, AJ, et al. β-Hydroxybutyrate is reduced in humans with obesity-related NAFLD and displays a dose-dependent effect on skeletal muscle mitochondrial respiration in vitro. Am J Physiology-Endocrinology Metab (2020) 319:E187–E195. doi:10.1152/ajpendo.00058.2020
24. McGarry, JD, and Foster, DW. Regulation of hepatic fatty acid oxidation and ketone body production. Annu Rev Biochem (1980) 49:395–420. doi:10.1146/annurev.bi.49.070180.002143
25. Hegardt, FG. Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: a control enzyme in ketogenesis. Biochem J (1999) 338(Pt 3):569–82. doi:10.1042/0264-6021:3380569
26. Laffel, L. Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab Res Rev (1999) 15:412–26. doi:10.1002/(sici)1520-7560(199911/12)15:6<412::aid-dmrr72>3.0.co;2-8
27. Wang, YH, Liu, CL, Chiu, WC, Twu, YC, and Liao, YJ. HMGCS2 mediates ketone production and regulates the proliferation and metastasis of hepatocellular carcinoma. Cancers (Basel) (2019) 11:1876. doi:10.3390/cancers11121876
28. Ding, R, Chen, T, Zhang, Y, Chen, X, Zhuang, L, and Yang, Z. HMGCS2 in metabolic pathways was associated with overall survival in hepatocellular carcinoma: a LASSO-derived study. Sci Prog (2021) 104:003685042110317. doi:10.1177/00368504211031749
29. Rojnueangnit, K, Maneechai, P, Thaweekul, P, Piriyanon, P, Khositseth, S, Ittiwut, C, et al. Expanding phenotypic and mutational spectra of mitochondrial HMG-CoA synthase deficiency. Eur J Med Genet (2020) 63:104086. doi:10.1016/j.ejmg.2020.104086
30. Ago, Y, Otsuka, H, Sasai, H, Abdelkreem, E, Nakama, M, Aoyama, Y, et al. Japanese patients with mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: in vitro functional analysis of five novel HMGCS2 mutations. Exp Ther Med (2020) 20:1. doi:10.3892/etm.2020.9166
31. Urganç, N, Arapoglu, M, Evruke, M, and Aydin, A. A rare cause of hepatomegaly: 3-hydroxy-3-methylglutaryl coenzyme-a lyase deficiency. J Pediatr Gastroenterol Nutr (2001) 33:339–41. doi:10.1097/00005176-200109000-00022
32. Arima, Y, Nakagawa, Y, Takeo, T, Ishida, T, Yamada, T, Hino, S, et al. Murine neonatal ketogenesis preserves mitochondrial energetics by preventing protein hyperacetylation. Nat Metab (2021) 3:196–210. doi:10.1038/s42255-021-00342-6
33. d'Avignon, DA, Puchalska, P, Ercal, B, Chang, Y, Martin, SE, Graham, MJ, et al. Hepatic ketogenic insufficiency reprograms hepatic glycogen metabolism and the lipidome. JCI Insight (2018) 3. doi:10.1172/jci.insight.99762
34. Cotter, DG, Ercal, B, Huang, X, Leid, JM, d’Avignon, DA, Graham, MJ, et al. Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J Clin Invest (2014) 124:5175–90. doi:10.1172/JCI76388
35. Mederacke, I, Hsu, CC, Troeger, JS, Huebener, P, Mu, X, Dapito, DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun (2013) 4:2823. doi:10.1038/ncomms3823
36. Pradere, JP, Kluwe, J, De Minicis, S, Jiao, JJ, Gwak, GY, Dapito, DH, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology (2013) 58:1461–73. doi:10.1002/hep.26429
37. Puchalska, P, Martin, SE, Huang, X, Lengfeld, JE, Daniel, B, Graham, MJ, et al. Hepatocyte-macrophage acetoacetate shuttle protects against tissue fibrosis. Cel Metab (2019) 29:383–98.e7. doi:10.1016/j.cmet.2018.10.015
38. van Hasselt, PM, Ferdinandusse, S, Monroe, GR, Ruiter, JP, Turkenburg, M, Geerlings, MJ, et al. Monocarboxylate transporter 1 deficiency and ketone utilization. N Engl J Med (2014) 371:1900–7. doi:10.1056/NEJMoa1407778
39. Luo, X, Li, Z, Chen, L, Zhang, X, Zhu, X, Wang, Z, et al. Monocarboxylate transporter 1 in the liver modulates high-fat diet-induced obesity and hepatic steatosis in mice. Metabolism (2023) 143:155537. doi:10.1016/j.metabol.2023.155537
40. Williamson, DH. Mechanisms for the regulation of ketogenesis. Proc Nutr Soc (1981) 40:93–8. doi:10.1079/pns19810014
41. Cotter, DG, Ercal, B, d'Avignon, DA, Dietzen, DJ, and Crawford, PA. Impairments of hepatic gluconeogenesis and ketogenesis in PPARalpha-deficient neonatal mice. Am J Physiol Endocrinol Metab (2014) 307:E176–185. doi:10.1152/ajpendo.00087.2014
42. Montagner, A, Polizzi, A, Fouché, E, Ducheix, S, Lippi, Y, Lasserre, F, et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut (2016) 65:1202–14. doi:10.1136/gutjnl-2015-310798
43. Qi, C, Zhu, Y, and Reddy, JK. Peroxisome proliferator-activated receptors, coactivators, and downstream targets. Cel Biochem Biophys (2000) 32:187–204. doi:10.1385/cbb:32:1-3:187
44. Sengupta, S, Peterson, TR, Laplante, M, Oh, S, and Sabatini, DM. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature (2010) 468:1100–4. doi:10.1038/nature09584
45. Feng, J, Qiu, S, Zhou, S, Tan, Y, Bai, Y, Cao, H, et al. mTOR: a potential New target in nonalcoholic fatty liver disease. Int J Mol Sci (2022) 23:9196. doi:10.3390/ijms23169196
46. Gao, Y, Sheng, X, Tan, D, Kim, S, Choi, S, Paudel, S, et al. Identification of histone lysine acetoacetylation as a dynamic post-translational modification regulated by HBO1. Adv Sci (2023) 10:e2300032. doi:10.1002/advs.202300032
47. Xie, Z, Zhang, D, Chung, D, Tang, Z, Huang, H, Dai, L, et al. Metabolic regulation of gene expression by histone lysine β-hydroxybutyrylation. Mol Cel (2016) 62:194–206. doi:10.1016/j.molcel.2016.03.036
48. Shimazu, T, Hirschey, MD, Newman, J, He, W, Shirakawa, K, Le Moan, N, et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science (2013) 339:211–4. doi:10.1126/science.1227166
49. Puchalska, P, and Crawford, PA. Metabolic and signaling roles of ketone bodies in health and disease. Annu Rev Nutr (2021) 41:49–77. doi:10.1146/annurev-nutr-111120-111518
50. Bendridi, N, Selmi, A, Balcerczyk, A, and Pirola, L. Ketone bodies as metabolites and signalling molecules at the crossroad between inflammation and epigenetic control of cardiometabolic disorders. Int J Mol Sci (2022) 23:14564. doi:10.3390/ijms232314564
51. Zhou, T, Cheng, X, He, Y, Xie, Y, Xu, F, Xu, Y, et al. Function and mechanism of histone β-hydroxybutyrylation in health and disease. Front Immunol (2022) 13:981285. doi:10.3389/fimmu.2022.981285
52. Hwang, CY, Choe, W, Yoon, KS, Ha, J, Kim, SS, Yeo, EJ, et al. Molecular mechanisms for ketone body metabolism, signaling functions, and therapeutic potential in cancer. Nutrients (2022) 14:4932. doi:10.3390/nu14224932
53. Asif, S, Morrow, NM, Mulvihill, EE, and Kim, KH. Understanding dietary intervention-mediated epigenetic modifications in metabolic diseases. Front Genet (2020) 11:590369. doi:10.3389/fgene.2020.590369
54. Xu, BT, Teng, F, Wu, Q, Wan, S, Li, X, Tan, X, et al. Bdh1 overexpression ameliorates hepatic injury by activation of Nrf2 in a MAFLD mouse model. Cell Death Discov (2022) 8:49. doi:10.1038/s41420-022-00840-w
55. Chen, Y, Ouyang, X, Hoque, R, Garcia-Martinez, I, Yousaf, MN, Tonack, S, et al. β-Hydroxybutyrate protects from alcohol-induced liver injury via a Hcar2-cAMP dependent pathway. J Hepatol (2018) 69:687–96. doi:10.1016/j.jhep.2018.04.004
56. Miyauchi, T, Uchida, Y, Kadono, K, Hirao, H, Kawasoe, J, Watanabe, T, et al. Up-regulation of FOXO1 and reduced inflammation by β-hydroxybutyric acid are essential diet restriction benefits against liver injury. Proc Natl Acad Sci U S A (2019) 116:13533–42. doi:10.1073/pnas.1820282116
57. Hazem, SH, Hamed, MF, Saad, MA, and Gameil, NM. Comparison of lactate and β-hydroxybutyrate in the treatment of concanavalin-A induced hepatitis. Int Immunopharmacology (2018) 61:376–84. doi:10.1016/j.intimp.2018.06.026
58. Moore, MP, Cunningham, RP, Davis, RAH, Deemer, SE, Roberts, BM, Plaisance, EP, et al. A dietary ketone ester mitigates histological outcomes of NAFLD and markers of fibrosis in high-fat diet fed mice. Am J Physiology-Gastrointestinal Liver Physiol (2021) 320:G564–G572. doi:10.1152/ajpgi.00259.2020
59. Rushing, KA, Bolyard, ML, Kelty, T, Wieschhaus, N, Pavela, G, Rector, RS, et al. Dietary ketone ester attenuates the accretion of adiposity and liver steatosis in mice fed a high-fat, high-sugar diet. Front Physiol (2023) 14:1165224. doi:10.3389/fphys.2023.1165224
60. Kim, KE, Jung, Y, Min, S, Nam, M, Heo, RW, Jeon, BT, et al. Caloric restriction of db/db mice reverts hepatic steatosis and body weight with divergent hepatic metabolism. Sci Rep (2016) 6:30111. doi:10.1038/srep30111
61. Ravussin, E, Redman, LM, Rochon, J, Das, SK, Fontana, L, Kraus, WE, et al. A 2-year randomized controlled trial of human caloric restriction: feasibility and effects on predictors of health span and longevity. The Journals Gerontol Ser A: Biol Sci Med Sci (2015) 70:1097–104. doi:10.1093/gerona/glv057
62. Lin, AL, Zhang, W, Gao, X, and Watts, L. Caloric restriction increases ketone bodies metabolism and preserves blood flow in aging brain. Neurobiol Aging (2015) 36:2296–303. doi:10.1016/j.neurobiolaging.2015.03.012
63. Ferguson, BS, Sahoo, P, McGrail, E, Francois, A, and Stratton, MS. Modestly increased incidence of ketosis in caloric restriction does not significantly alter the effects of caloric restriction. The J Nutr Health Aging (2022) 26:657–62. doi:10.1007/s12603-022-1815-7
64. Ravaut, G, Carneiro, A, and Mounier, C. Exploring the impacts of ketogenic diet on reversible hepatic steatosis: initial analysis in male mice. Front Nutr (2024) 11:1290540. doi:10.3389/fnut.2024.1290540
65. Luukkonen, PK, Dufour, S, Lyu, K, Zhang, XM, Hakkarainen, A, Lehtimäki, TE, et al. Effect of a ketogenic diet on hepatic steatosis and hepatic mitochondrial metabolism in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A (2020) 117:7347–54. doi:10.1073/pnas.1922344117
66. Rector, RS, Uptergrove, GM, Morris, EM, Borengasser, SJ, Laughlin, MH, Booth, FW, et al. Daily exercise vs. caloric restriction for prevention of nonalcoholic fatty liver disease in the OLETF rat model. Am J Physiology-Gastrointestinal Liver Physiol (2011) 300:G874–883. doi:10.1152/ajpgi.00510.2010
67. Dong, F, Zhang, Y, Huang, Y, Wang, Y, Zhang, G, Hu, X, et al. Long-term lifestyle interventions in middle-aged and elderly men with nonalcoholic fatty liver disease: a randomized controlled trial. Sci Rep (2016) 6:36783. doi:10.1038/srep36783
68. Mardinoglu, A, Wu, H, Bjornson, E, Zhang, C, Hakkarainen, A, Räsänen, SM, et al. An integrated understanding of the rapid metabolic benefits of a carbohydrate-restricted diet on hepatic steatosis in humans. Cel Metab (2018) 27:559–71.e5. doi:10.1016/j.cmet.2018.01.005
69. Tendler, D, Lin, S, Yancy, WS, Mavropoulos, J, Sylvestre, P, Rockey, DC, et al. The effect of a low-carbohydrate, ketogenic diet on nonalcoholic fatty liver disease: a pilot study. Dig Dis Sci (2007) 52:589–93. doi:10.1007/s10620-006-9433-5
70. Newman, JC, Covarrubias, AJ, Zhao, M, Yu, X, Gut, P, Ng, CP, et al. Ketogenic diet reduces midlife mortality and improves memory in aging mice. Cel Metab (2017) 26:547–57.e8. doi:10.1016/j.cmet.2017.08.004
71. Cunha, GM, Guzman, G, Correa De Mello, LL, Trein, B, Spina, L, Bussade, I, et al. Efficacy of a 2-month very low-calorie ketogenic diet (VLCKD) compared to a standard low-calorie diet in reducing visceral and liver fat accumulation in patients with obesity. Front Endocrinol (Lausanne) (2020) 11:607. doi:10.3389/fendo.2020.00607
72. Nasser, S, Solé, T, Vega, N, Thomas, T, Balcerczyk, A, Strigini, M, et al. Ketogenic diet administration to mice after a high-fat-diet regimen promotes weight loss, glycemic normalization and induces adaptations of ketogenic pathways in liver and kidney. Mol Metab (2022) 65:101578. doi:10.1016/j.molmet.2022.101578
73. Guo, W, Cao, H, Shen, Y, Li, W, Wang, W, Cheng, L, et al. Role of liver FGF21-KLB signaling in ketogenic diet-induced amelioration of hepatic steatosis. Nutr Diabetes (2024) 14:18. doi:10.1038/s41387-024-00277-3
74. Badman, MK, Pissios, P, Kennedy, AR, Koukos, G, Flier, JS, and Maratos-Flier, E. Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cel Metab (2007) 5:426–37. doi:10.1016/j.cmet.2007.05.002
75. Newman, JC, and Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol and Metab (2014) 25:42–52. doi:10.1016/j.tem.2013.09.002
76. Long, F, Bhatti, MR, Kellenberger, A, Sun, W, Modica, S, Höring, M, et al. A low-carbohydrate diet induces hepatic insulin resistance and metabolic associated fatty liver disease in mice. Mol Metab (2023) 69:101675. doi:10.1016/j.molmet.2023.101675
77. Wei, SJ, Schell, JR, Chocron, ES, Varmazyad, M, Xu, G, Chen, WH, et al. Ketogenic diet induces p53-dependent cellular senescence in multiple organs. Sci Adv (2024) 10:eado1463. doi:10.1126/sciadv.ado1463
78. Di Francesco, A, Di Germanio, C, Bernier, M, and de Cabo, R. A time to fast. Science (2018) 362:770–5. doi:10.1126/science.aau2095
79. Longo, VD, and Mattson, MP. Fasting: molecular mechanisms and clinical applications. Cel Metab (2014) 19:181–92. doi:10.1016/j.cmet.2013.12.008
80. Geisler, CE, Ghimire, S, Bogan, RL, and Renquist, BJ. Role of ketone signaling in the hepatic response to fasting. Am J Physiology-Gastrointestinal Liver Physiol (2019) 316:G623–G631. doi:10.1152/ajpgi.00415.2017
81. Gallage, S, Ali, A, Barragan Avila, JE, Seymen, N, Ramadori, P, Joerke, V, et al. A 5:2 intermittent fasting regimen ameliorates NASH and fibrosis and blunts HCC development via hepatic PPARalpha and PCK1. Cell Metab (2024) 36:1371–93 e1377. doi:10.1016/j.cmet.2024.04.015
82. Elsayed, HRH, El-Nablaway, M, Khattab, BA, Sherif, RN, Elkashef, WF, Abdalla, AM, et al. Independent of calorie intake, short-term alternate-day fasting alleviates NASH, with modulation of markers of lipogenesis, autophagy, apoptosis, and inflammation in rats. J Histochem Cytochem (2021) 69:575–96. doi:10.1369/00221554211041607
83. Marinho, Td. S, Ornellas, F, Barbosa-da-Silva, S, Mandarim-de-Lacerda, CA, and Aguila, MB. Beneficial effects of intermittent fasting on steatosis and inflammation of the liver in mice fed a high-fat or a high-fructose diet. Nutrition (2019) 65:103–12. doi:10.1016/j.nut.2019.02.020
84. Kim, KE, Shin, HJ, Ju, Y, Jung, Y, An, HS, Lee, SJ, et al. Intermittent fasting attenuates metabolic-dysfunction-associated steatohepatitis by enhancing the hepatic autophagy-lysosome pathway. Nutrients (2023) 15:4574. doi:10.3390/nu15214574
85. Damasceno de Lima, R, Fudoli Lins Vieira, R, Rosetto Muñoz, V, Chaix, A, Azevedo Macedo, AP, Calheiros Antunes, G, et al. Time-restricted feeding combined with resistance exercise prevents obesity and improves lipid metabolism in the liver of mice fed a high-fat diet. Am J Physiology-Endocrinology Metab (2023) 325:E513–E528. doi:10.1152/ajpendo.00129.2023
86. Wei, X, Lin, B, Huang, Y, Yang, S, Huang, C, Shi, L, et al. Effects of time-restricted eating on nonalcoholic fatty liver disease: the TREATY-FLD randomized clinical trial. JAMA Netw Open (2023) 6:e233513. doi:10.1001/jamanetworkopen.2023.3513
87. Fu, ZD, Cai, XL, Yang, WJ, Zhao, MM, Li, R, and Li, YF. Novel glucose-lowering drugs for non-alcoholic fatty liver disease. World J Diabetes (2021) 12:84–97. doi:10.4239/wjd.v12.i1.84
88. Fougerat, A, Montagner, A, Loiseau, N, Guillou, H, and Wahli, W. Peroxisome proliferator-activated receptors and their novel ligands as candidates for the treatment of non-alcoholic fatty liver disease. Cells (2020) 9:1638. doi:10.3390/cells9071638
89. Zou, W, Zhang, C, Gu, X, Li, X, and Zhu, H. Metformin in combination with malvidin prevents progression of non-alcoholic fatty liver disease via improving lipid and glucose metabolisms, and inhibiting inflammation in type 2 diabetes rats. Drug Des Develop Ther (2021) 15:2565–76. doi:10.2147/DDDT.S307257
90. Pinyopornpanish, K, Leerapun, A, Pinyopornpanish, K, and Chattipakorn, N. Effects of metformin on hepatic steatosis in adults with nonalcoholic fatty liver disease and diabetes: insights from the cellular to patient levels. Gut and Liver (2021) 15:827–40. doi:10.5009/gnl20367
91. Tessari, P, and Tiengo, A. Metformin treatment of rats with diet-induced overweight and hypertriglyceridemia decreases plasma triglyceride concentrations, while decreasing triglyceride and increasing ketone body output by the isolated perfused liver. Acta Diabetol (2008) 45:143–5. doi:10.1007/s00592-008-0032-0
92. Cuyas, E, Fernández-Arroyo, S, Buxó, M, Pernas, S, Dorca, J, Álvarez, I, et al. Metformin induces a fasting- and antifolate-mimicking modification of systemic host metabolism in breast cancer patients. Aging (Albany NY) (2019) 11:2874–88. doi:10.18632/aging.101960
93. Qiu, YY, Zhang, J, Zeng, FY, and Zhu, YZ. Roles of the peroxisome proliferator-activated receptors (PPARs) in the pathogenesis of nonalcoholic fatty liver disease (NAFLD). Pharmacol Res (2023) 192:106786. doi:10.1016/j.phrs.2023.106786
94. Souza-Mello, V. Peroxisome proliferator-activated receptors as targets to treat non-alcoholic fatty liver disease. World J Hepatol (2015) 7:1012–9. doi:10.4254/wjh.v7.i8.1012
95. Shinozaki, S, Tahara, T, Miura, K, Kawarai Lefor, A, and Yamamoto, H. Pemafibrate therapy for non-alcoholic fatty liver disease is more effective in lean patients than obese patients. Clin Exp Hepatol (2022) 8:278–83. doi:10.5114/ceh.2022.120099
96. Honda, Y, Kessoku, T, Ogawa, Y, Tomeno, W, Imajo, K, Fujita, K, et al. Pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator, improves the pathogenesis in a rodent model of nonalcoholic steatohepatitis. Sci Rep (2017) 7:42477. doi:10.1038/srep42477
97. Bian, H, Liu, YM, and Chen, ZN. New avenues for NASH therapy by targeting ACC. Cel Metab (2022) 34:191–3. doi:10.1016/j.cmet.2022.01.001
98. Alkhouri, N, Lawitz, E, Noureddin, M, DeFronzo, R, and Shulman, GI. GS-0976 (Firsocostat): an investigational liver-directed acetyl-CoA carboxylase (ACC) inhibitor for the treatment of non-alcoholic steatohepatitis (NASH). Expert Opin Investig Drugs (2020) 29:135–41. doi:10.1080/13543784.2020.1668374
99. Calle, RA, Amin, NB, Carvajal-Gonzalez, S, Ross, TT, Bergman, A, Aggarwal, S, et al. ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: two parallel, placebo-controlled, randomized phase 2a trials. Nat Med (2021) 27:1836–48. doi:10.1038/s41591-021-01489-1
100. Zhang, XJ, Ji, YX, Cheng, X, Cheng, Y, Yang, H, Wang, J, et al. A small molecule targeting ALOX12-ACC1 ameliorates nonalcoholic steatohepatitis in mice and macaques. Sci Transl Med (2021) 13:eabg8116. doi:10.1126/scitranslmed.abg8116
101. Kim, CW, Addy, C, Kusunoki, J, Anderson, NN, Deja, S, Fu, X, et al. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cel Metab (2017) 26:394–406.e6. doi:10.1016/j.cmet.2017.07.009
102. Gulette, GA, Hass, DT, Pandey, K, Zhang, Q, Han, JY, Engel, A, et al. Reassessing retinal pigment epithelial ketogenesis: enzymatic assays for ketone body levels provide inaccurate results. Exp Eye Res (2024) 245:109966. doi:10.1016/j.exer.2024.109966
103. Hsia, DS, Grove, O, and Cefalu, WT. An update on sodium-glucose co-transporter-2 inhibitors for the treatment of diabetes mellitus. Curr Opin Endocrinol Diabetes and Obes (2017) 24:73–9. doi:10.1097/MED.0000000000000311
104. Yaribeygi, H, Maleki, M, Butler, AE, Jamialahmadi, T, and Sahebkar, A. New insights into cellular links between sodium-glucose cotransporter-2 inhibitors and ketogenesis. J Cell Biochem (2022) 123:1879–90. doi:10.1002/jcb.30327
105. Hoong, CWS, and Chua, MWJ. SGLT2 inhibitors as calorie restriction mimetics: insights on longevity pathways and age-related diseases. Endocrinology (2021) 162. doi:10.1210/endocr/bqab079
106. Wei, Q, Xu, X, Guo, L, Li, J, and Li, L. Effect of SGLT2 inhibitors on type 2 diabetes mellitus with non-alcoholic fatty liver disease: a meta-analysis of randomized controlled trials. Front Endocrinol (Lausanne) (2021) 12:635556. doi:10.3389/fendo.2021.635556
107. Capozzi, ME, Coch, RW, Koech, J, Astapova, II, Wait, JB, Encisco, SE, et al. The limited role of glucagon for ketogenesis during fasting or in response to SGLT2 inhibition. Diabetes (2020) 69:882–92. doi:10.2337/db19-1216
108. Berger, JH, Matsuura, TR, Bowman, CE, Taing, R, Patel, J, Lai, L, et al. Sodium-glucose co-transporter 2 inhibitors act independently of SGLT2 to confer benefit for heart failure with reduced ejection fraction in mice. bioRxiv (2024) 2004.2029:591665. doi:10.1101/2024.04.29.591665
109. Al Batran, R, Gopal, K, Capozzi, ME, Chahade, JJ, Saleme, B, Tabatabaei-Dakhili, SA, et al. Pimozide alleviates hyperglycemia in diet-induced obesity by inhibiting skeletal muscle ketone oxidation. Cel Metab (2020) 31:909–19.e8. doi:10.1016/j.cmet.2020.03.017
110. Uehara, K, Sostre-Colón, J, Gavin, M, Santoleri, D, Leonard, KA, Jacobs, RL, et al. Activation of liver mTORC1 protects against NASH via dual regulation of VLDL-TAG secretion and de novo lipogenesis. Cell Mol Gastroenterol Hepatol (2022) 13:1625–47. doi:10.1016/j.jcmgh.2022.02.015
111. Musso, G, Saba, F, Cassader, M, and Gambino, R. Diabetic ketoacidosis with SGLT2 inhibitors. BMJ (2020) 371:m4147. doi:10.1136/bmj.m4147
112. Slade, S, and Ashurst, J. Diet-induced ketoacidosis in a non-diabetic: a case report. Clin Pract Cases Emerg Med (2020) 4:259–62. doi:10.5811/cpcem.2020.2.44736
113. Smolensky, I, Zajac-Bakri, K, Odermatt, TS, Brégère, C, Cryan, JF, Guzman, R, et al. Sex-specific differences in metabolic hormone and adipose tissue dynamics induced by moderate low-carbohydrate and ketogenic diet. Sci Rep (2023) 13:16465. doi:10.1038/s41598-023-43587-9
114. Chaix, A, Deota, S, Bhardwaj, R, Lin, T, and Panda, S. Sex- and age-dependent outcomes of 9-hour time-restricted feeding of a Western high-fat high-sucrose diet in C57BL/6J mice. Cel Rep (2021) 36:109543. doi:10.1016/j.celrep.2021.109543
Keywords: MASLD, ketone bodies, ketogenesis, dietary interventions, pharmacological interventions
Citation: Kwon S, Jeyaratnam R and Kim K-H (2024) Targeting ketone body metabolism to treat fatty liver disease. J. Pharm. Pharm. Sci 27:13375. doi: 10.3389/jpps.2024.13375
Received: 12 June 2024; Accepted: 13 September 2024;
Published: 30 September 2024.
Edited by:
John Reyes Ussher, University of Alberta, CanadaCopyright © 2024 Kwon, Jeyaratnam and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kyoung-Han Kim, aGtpbUBvdHRhd2FoZWFydC5jYQ==