Abstract
In xenotransplantation, the vascular endothelium serves as the first point of contact between the recipient’s blood and the transplanted donor organ. The loss of the endothelium’s ability to control the plasma cascades plays a critical role in the dysregulation of the complement and coagulation systems, which greatly contribute to graft rejection and hinder long-term xenograft survival. Although it is known that an intact glycocalyx is a key feature of a resting endothelium that exhibits optimal anticoagulant and anti-inflammatory properties, the role of the endothelial glycocalyx in xenotransplantation is barely investigated so far. Here, we discuss the central role of endothelial cells and the sugar-rich endothelial glycocalyx in regulating the plasma cascades, and how the loss of these functions contributes to graft damage and rejection. We highlight the importance of preserving the regulatory functions of both endothelial cells and the glycocalyx as strategies to improve xenotransplantation outcomes.
Introduction
The luminal side of the vascular endothelium is lined by a monolayer of endothelial cells (ECs), which function far beyond merely acting as the barrier between blood and tissue. EC functions include regulating the vascular tone, mechanotransduction, permeability, oxygen and nutrient supply, vascular hemostasis, as well as immunomodulatory activities [1–6]. The outermost layer of ECs is covered by the endothelial glycocalyx, a sugar-rich layer composed of diverse sugar conjugates, such as glycoproteins, proteoglycans, and glycolipids, which serve as the first point of contact for cellular and humoral components of the immune system with the endothelium [7–10].
The well-balanced control of the plasma cascade systems – complement, coagulation, fibrinolysis, and kallikrein/kinin – by ECs is crucial in healthy and pathological conditions, including pig-to-human xenotransplantation [11]. However, this control is lost due to incompatibilities between regulatory factors provided by the porcine ECs and the human plasma components present in the recipient’s blood, ultimately leading to graft damage and rejection [12–14]. The activation of the xenograft ECs and subsequent loss of the regulatory control of the plasma cascades have been shown to be the significant events leading to organ failure and rejection [15–19].
Genetic modifications have been used to overcome xenorejection due to humoral responses by eliminating the synthesis of xenoantigens [20–22]. In addition, the expression of human complement and coagulation regulatory genes in donor pigs has been used to regain control over the activation of the plasma cascade systems [23–26]. While impressive progress has been achieved, complement and coagulation dysregulations are still presented as key features of short- or long-term xenograft rejection in preclinical and clinical models [26–30]. This indicates that the currently available strategies to preserve the regulatory functions of xenograft ECs are not sufficient to achieve long-lasting control and graft survival.
In this review we outline the critical roles of ECs and the glycocalyx in regulating the plasma cascade systems. We also highlight the interaction of the EC glycocalyx with the recipient’s plasma cascades as well as its role in xenograft rejection. Finally, we discuss the potential of EC glycocalyx protection as a possible therapeutic strategy to improve graft survival and function.
Regulation of Plasma Cascades by Endothelial Cells
Normal ECs are typically in a quiescent state, displaying minimal proliferation, migration, and permeability. The surface of healthy ECs is also in an anticoagulant, anti-inflammatory, and pro-fibrinolytic state. The maintenance of this quiescent state is achieved by complex and active functions involving the continuous production and regulation of various molecules and receptors [1, 11].
Coagulation and Fibrinolysis
Coagulation and fibrinolysis are essential physiological processes that occur continuously, maintaining the delicate equilibrium between fibrin formation and breakdown in the plasma [31]. Activation of the coagulation cascade can be initiated via two pathways: the intrinsic pathway, also called the contact activation pathway, and the extrinsic pathway, which is triggered by tissue factor (TF). Both pathways result in the activation of factor X and subsequent production of thrombin, a serine protease that converts fibrinogen into fibrin, ultimately leading to clot formation. The extrinsic pathway mainly serves as a mechanism for hemostatic control and response to injury, while the activation of the intrinsic pathway has been associated with pathological clotting [32, 33].
Under normal physiological conditions, ECs prevent thrombus formation by providing a non-adhesive surface to prevent the activation of platelets and the coagulation cascade. More importantly, ECs are actively preventing thrombus formation by expressing soluble and membrane-bound molecules with anticoagulant properties [34]. ECs produce soluble molecules such as nitric oxide (NO) and prostacyclin (PGI2) which inhibit platelet aggregation [35–37]. To inhibit coagulation via the TF pathway, ECs also express tissue factor pathway inhibitor (TFPI) [38–40]. Furthermore, as the formation of fibrin happens continuously in the plasma, ECs prevent the accumulation of fibrin by secreting tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (u-PA) [41, 42]. These two molecules will convert plasminogen to the active plasmin, which breaks down fibrin clots accumulating on the EC surface. To further enhance its anticoagulant properties, ECs bind inhibitors of coagulation factors, preventing the progression of the coagulation cascade. For instance, heparan sulfate (HS), present on the EC glycocalyx, bind the liver-derived antithrombin III (ATIII), which inhibits coagulation by preventing thrombin and other coagulation factors from binding to their substrates [43–45]. Moreover, the membrane-bound thrombomodulin (TBM) expressed on the EC surface can bind circulating thrombin and inhibit its procoagulant function, altering thrombin’s affinity from binding fibrinogen to binding and activating anticoagulant protein C [34, 46, 47]. ECs also express a high affinity receptor for protein C (EPCR), which binds protein C and further enhances its TBM-thrombin complex-mediated activation [48]. The coagulation inhibitory properties of ECs are summarized in (Table 1).
TABLE 1
| Molecule | Form | Target action | Function | Ref |
|---|---|---|---|---|
| Nitric Oxide (NO) | Soluble gas | Platelets | Diffuses to nearby platelets and prevents calcium release, which ultimately prevents platelet activation and aggregation | [36, 37] |
| Prostacyclin (PGI2) | Soluble protein | Platelets | Inhibition of platelet activation through calcium release inhibition | [35] |
| Heparane Sulfate (HS) | Membrane-bound glycosami noglycans | ATIII | Bound ATIII inhibits thrombin (factor II) and other serine proteases binding to their substrates | [43, 44] |
| Tissue Factor Pathway Inhibitor (TFPI) | Soluble protein | Tissue factor (Factor III) | Limits the TF (factor III) activity which initiates prothrombin transformation into thrombin | [38, 39] |
| Thrombo modulin (TBM) | Membrane-bound protein | Thrombin, protein C | Binds to thrombin and reduces the ability of thrombin to convert fibrinogen into fibrin TBM-thrombin complex enhances protein C activity to inhibit coagulation factors Va and VIIIa |
[46, 48] |
| t-PA | Soluble protein | Plasminogen | Activates plasminogen into plasmin and initiates fibrinolysis | [31, 41] |
| u-PA | Soluble protein | Plasminogen | Activates plasminogen into plasmin and initiates fibrinolysis | [31, 41] |
| EPCR | Membrane-bound protein | Protein C | Binds and promotes protein C activation by presenting protein C to TBM-thrombin complex, ultimately inhibiting factor Va and VIIIa | [48] |
Anticoagulant properties of endothelial cells.
Complement Cascade
The complement system plays a crucial role in clearing immune complexes and injured cells. It can be activated via three different activation pathways, all of which converge at the C3 level. Activation of the cascades produces opsonins (C3b, C4b), which mark the targets for subsequent removal, and the membrane attack complex (MAC, C5b-9), which directly lyses the target cells. Additionally, complement activation generates the anaphylatoxins C3a and C5a, promoting leukocyte recruitment and inflammation. However, the activation of the complement system acts as a double-edged sword because the effectors of complement activation have the potential to harm the host. To prevent this, the complement system is highly regulated through the expression of soluble and membrane-bound molecules to avoid the undesired effect of complement activation. ECs, as the first layer constantly exposed to the complement mediators in the plasma, play an indispensable role in this regulatory process.
ECs express membrane-bound components and secrete soluble molecules that prevent the activation of the complement cascade or deposition of complement activation products (Table 2). ECs express the surface molecules CD46 and CD55, which inhibit the activation of complement pathways, and CD59, which prevents the formation of MAC on the surface of ECs [50, 51, 54]. ECs also secrete C1 inhibitor (C1-INH), clusterin, factor H, and factor I, which prevent the formation of complement effectors at various stages [49, 52, 53, 55, 56].
TABLE 2
| Molecule | Form | Target action | Function | Ref |
|---|---|---|---|---|
| C1 inhibitor | Soluble protein | C1 | Inhibits the activation of complement C1 | [49] |
| CD46 (MCP) | Membrane-bound protein | C3, C4 | Mediates the cleavage of C3b and C4b | [50] |
| CD55 (DAF) | Membrane-bound protein | C3 convertase C5 convertase |
Destabilizes C3/C5 convertases in the classical and the alternative pathways | [51] |
| Factor H | Soluble protein | C3b | Inactivates the C3 and C5 convertases | [52] |
| Factor I | Soluble protein | C3b, C4b | Cofactor for the C3b and C4b cleavage by factor H | [53] |
| CD59 | Membrane-bound protein | C5b-9 | Prevents C5b-9 formation | [54] |
| Clusterin | Soluble protein | C5b-9 | Prevents C5b-9 formation | [55, 56] |
Complement regulatory properties of endothelial cells.
Both the complement and coagulation systems comprise of serine proteases with common ancestral genes [57, 58]. There is substantial evidence of crosstalk between complement and coagulation factors, leading to mutual engagement of both systems. For instance, proteases such as thrombin, factor XII (Hageman factor), and plasmin play roles in both coagulation and complement activation [59–62]. This crosstalk also influences the regulation of the plasma cascade pathways by ECs through shared complement-coagulation regulators such as C1-INH, TBM, TFPI, CD46, CD55, and CD59 [58, 63–65]. Consequently, a lack of function of the previously mentioned inhibitors, can also contribute to concurrent activation and amplification of complement and coagulation pathways under pathological conditions, resulting in excessive activation of both systems [64, 66, 67].
Genetic Modification Strategies to Overcome Complement and Coagulation Dysregulation in Xenotransplantation
In xenotransplantation, interaction of the porcine ECs with the human plasma cascades presents great immunological challenges due to the presence of xenoantigens on porcine ECs, and molecular incompatibilities between porcine plasma cascade regulators and human plasma cascade components [21, 22, 68]. The porcine glycocalyx presents sugars carrying α-Gal, N-glycolylneuraminic acid (Neu5Gc), and Sda epitopes, which can be recognized as foreign by natural antibodies circulating in human blood, and have been shown to play a significant role in the immune response to pig grafts in xenotransplantation [22, 68–71]. Antibody-mediated complement activation will lead to hyperacute rejection, a rapid destruction of the xenograft due to the complement attack, accompanied by interstitial hemorrhage, edema, and microvascular thrombosis [21, 72, 73]. The lack of complement and coagulation protection on the surface of ECs due to the interspecies molecular incompatibilities also leads to complement-mediated injury and coagulopathy, which are evident in various stages of xenograft rejection [74–76].
The first approach to avoid activation of complement on the EC surface in xenotransplantation is the deletion of porcine xenoantigens, which was shown to prevent hyperacute rejection [20, 24, 70, 77]. Another approach to provide better control of the plasma cascades by the endothelium is the transgenic overexpression of human complement and coagulation regulatory factors [16, 23–26]. Strategies to overcome the complement and coagulation dysregulation are illustrated in Figure 1. Genetically modified pigs carrying single or multiple xenoantigens knockouts and expressing varying combination of plasma cascade regulatory proteins have been developed and show different survival rates [26, 81, 87–89].
FIGURE 1
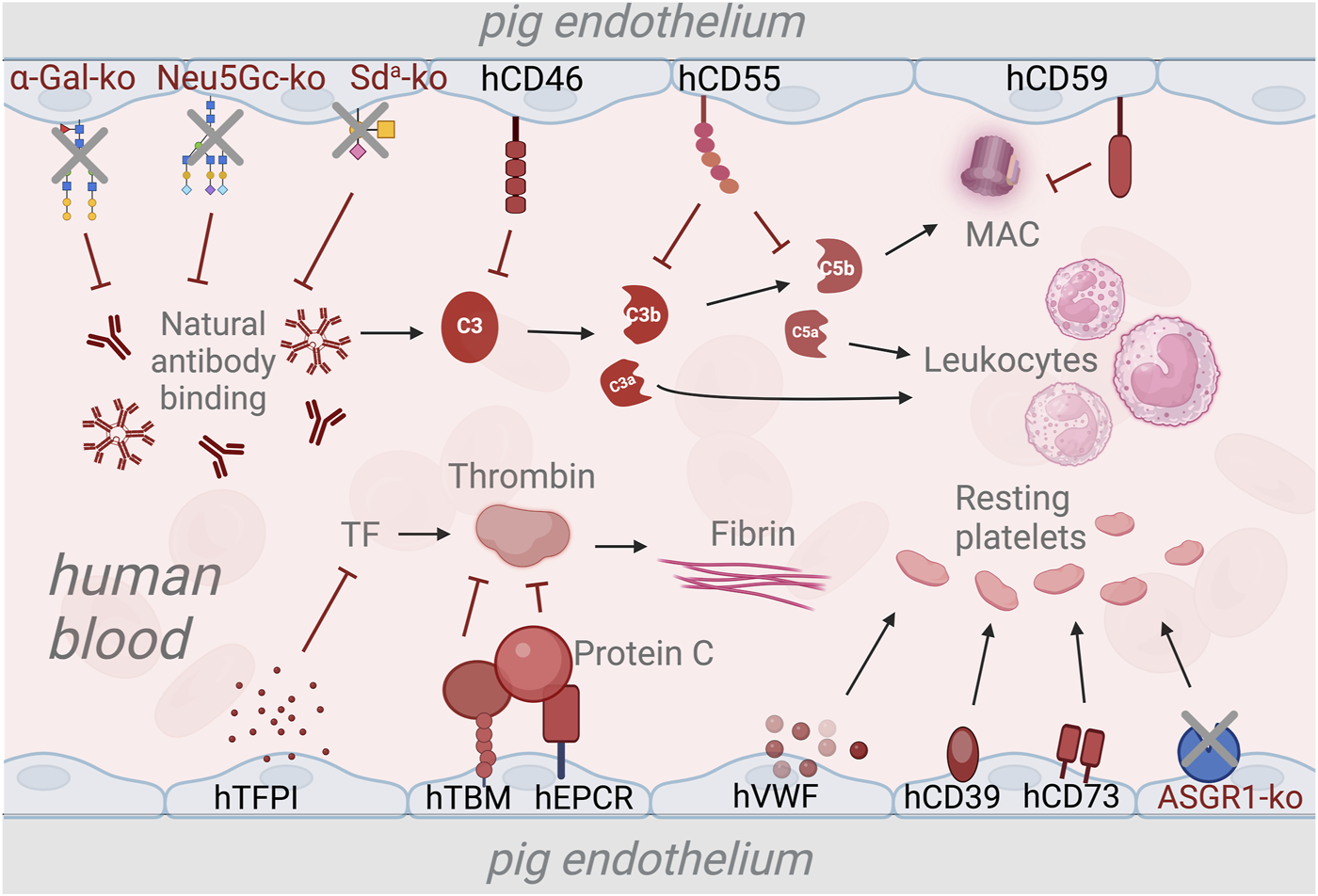
Strategies to overcome the complement and coagulation dysregulation in xenotransplantation using genetic modifications. The deletion of genes responsible for xenoantigen epitopes prevents the binding of natural antibodies and subsequent activation of the complement system [20, 21, 24]. Transgenic expression of human complement regulatory genes such as human CD46 (hCD46), hCD55, and hCD59 inhibits the activation of the complement cascade and the formation of membrane attack complex (MAC) [75, 78, 79]. The expression of human tissue pathway inhibitor (hTFPI) is used to prevent activation of the coagulation cascade via the tissue factor (TF) pathway [80]. Additionally, Human thrombomodulin (hTBM) and endothelial protein C-receptor (hEPCR) are expressed to inhibit thrombin activation, thus preventing fibrin formation [26, 81]. To maintain the platelets in a resting state, transgenic expression of human von Willenbrand Factor (hVWF), triphosphate diphosphohydrolase-1 (human ENTPDase-1 or hCD39) and human CD73 [82–84]. Deletion of the asialoglycoprotein receptor-1 (ASGR1) gene is used as a strategy to reduce human platelet destruction by pig livers [85, 86].
The Role of the Endothelial Glycocalyx in Plasma Cascade Regulation
The function of ECs, from mechanotransduction, maintenance of vascular integrity and vascular tone, as well as regulating the interaction of EC with the immune components of the blood, is profoundly influenced by their glycocalyx component [90]. While the term glycocalyx broadly refers to surface sugar layer of any cells, the EC glycocalyx specifically refers to the carbohydrate-rich layer on the luminal side of ECs. The thickness and the diversity of the EC glycocalyx is influenced by the shear stress, plasma proteins, and extrinsic glycosylation factors in the blood [3, 10, 91–93]. The EC glycocalyx is made up of diverse sugar conjugates, including proteoglycans, glycoproteins, and glycolipids, facilitating the interaction of ECs with various immune mediators of the plasma cascades.
Proteoglycans are glycoproteins comprising a core protein (e.g., syndecan, glypican, and perlecan) that carries covalently attached linear polysaccharide glycosaminoglycans (GAGs). The main components of the EC glycocalyx GAGs are HS, chondroitin sulfate (CS) and dermatan sulfate (DS) [10, 94]. The EC glycocalyx also contains the non-sulfated GAG hyaluronan (HA), typically anchored by the CD44 receptor, or as a soluble component [95, 96]. GAGs are the most abundant sugars in the EC glycocalyx, contributing significantly to its molecular mass and negative charge, creating a transendothelial gradient for fluid transit, masking the adhesion molecules, and capturing circulating plasma proteins [97, 98]. GAGs are also essential in maintaining the quiescent state of ECs. They act as mechanotransducers, signaling to maintain EC function via the production of NO [90, 99]. GAGs can bind and modulate the activity of several growth factors, antiangiogenic, and inflammatory molecules such as VEGF, FGF, IL8, MMPs, angiopoietin-1, thrombospondin-1, endostatin, and PAI-1 [100–105]. Additionally, GAGs have binding sites for xantine-oxydoreductase (XOD) and endothelial superoxide dismutase (eSOD), preventing oxidative damage to ECs [106, 107].
HS is one of the most extensively studied GAGs and builds up most of the sugar components of the EC glycocalyx. HS is known for its critical role in various biological functions, from serving as a binding domain for signal transduction molecules to regulating the plasma cascade and immune interactions [108]. Plasma proteins bind to HS via their heparin-binding domain. This binding is specific, and through the differential expression of HS, ECs can selectively determine which plasma proteins will bind to the surface [109, 110]. HS on the EC glycocalyx can bind and potentiate the activity of complement and coagulation regulatory factors, including factor H, C1-INH, and ATIII, providing an anti-inflammatory and anticoagulant surface [44, 111–115]. Binding of growth factors and cytokines such as VEGF, FGF, and IL8 is predominantly facilitated by HS [43, 103, 116, 117]. HS also serves as a binding site for P-selectin and L-selectin, potentially acting as a decoy receptor to influence cell adhesion dynamics [118, 119]. Moreover, HS is also involved in the regulation of the kinin-kallikrein system through its capacity to recruit kinin precursors and mediate the activation of high molecular weight kininogen [120, 121].
The terminal glycans of mammalian glycoproteins and glycolipids are typically substituted by the negatively charged sialic acid, which plays an essential role in physiological and pathological processes, including complement, coagulation, and inflammation [72, 122–124]. Many important receptors and regulatory proteins on the EC surface carry N- and O-linked oligosaccharide chains and are classified as glycoproteins [94, 125]. The interaction between ECs and leukocytes is controlled by adhesion molecules such as siglecs and selectin receptors, that are heavily influenced by the cellular glycosylation patterns of these proteins [126, 127]. Factor H can bind to sialic acid, providing a protective layer on ECs and shielding the cells from the deposition of complement [128–131]. Glycosphingolipids (GSLs), a diverse group of glycolipids containing one or more glycans anchored to a ceramide, are known to play important roles in many cellular processes, such as signal transduction, cell-cell interactions, and immune response regulation, including the complement and coagulation systems [132–135]. While the role of GSLs has been extensively studied in neurodegenerative diseases, their role in the context of xenotransplantation immunology is less well-understood.
Glycocalyx Shedding
Due to its pivotal role in maintaining the normal function of ECs, destruction of the EC glycocalyx will lead to impairment of EC function and overall vascular hemostasis. Glycocalyx shedding is observed when ECs are activated and exhibit a pro-inflammatory state, including in trauma-related injury, ischemia/reperfusion injury, hemorrhagic shock, and hyperglycemia [10, 136–139]. In xenotransplantation, glycocalyx shedding associated with complement and coagulation activation after xenogeneic treatment has been observed in vitro and in vivo [140–144]. Disruption of the EC glycocalyx may hamper its mechanotransduction ability and induce edema due to elevated vascular permeability [145–149]. Shedding of the EC glycocalyx will result in the loss of anticoagulant and anti-inflammatory properties of the EC, leading to increased coagulation, decreased pro-fibrinolytic activity, increased complement deposition, and adhesion of leukocytes to the EC surface [138–140, 150].
Shedding of the EC glycocalyx during inflammation can be mediated by various glycocalyx modifying factors such as reactive oxygen species (ROS), matrix metalloproteinases (MMPs), and glycan-degrading enzymes (heparanase, hyaluronase, sulfatase, sialidase, etc.) [9, 10, 139, 151]. Other proteins with enzymatic functions such as thrombin, elastase, proteinase 3, plasminogen, as well as cathepsin B also contribute to degradation of the glycocalyx [152–154]. In cardiovascular diseases associated with vascular inflammation, the observed glycocalyx shedding can be mediated by multiple glycocalyx-degrading mediators [139, 155, 156].
Interaction of the Porcine Glycocalyx With Human Plasma Proteins in Xenotransplantation
One significant concern, and barely studied so far, are the possible differences in the binding of human plasma proteins to the human and porcine glycocalyx (Figure 2). The absence of glycans containing α-Gal, Neu5Gc, and Sda epitopes in humans highlights the species-specific differences in sugar synthesis and expression, as well as the disparity in glycocalyx composition between humans and pigs [21]. Due to its different composition, the porcine glycocalyx may have varying structure and affinities for human plasma cascade regulators such as ATIII, factor H, and C1-INH, which could reduce the anti-inflammatory and anticoagulant properties of the xenografted EC surface once a porcine organ has been grafted into a human recipient. Discrepancies in the binding affinities to human growth factors, cytokines, and chemokines could likely also cause a systemic effect due to loss of control of dissociated and active molecules by ECs [103, 109, 158]. Similarly, possible differences in enzyme kinetics and affinities between the porcine glycocalyx and glycocalyx-modifying enzymes could also affect the glycocalyx dynamics and function. Therefore, despite the effort to substitute the interspecies complement and coagulation incompatibility on a protein level in xenotransplantation, EC function in controlling the plasma cascade regulation might still not be optimal without proper functioning of the glycocalyx [144].
FIGURE 2
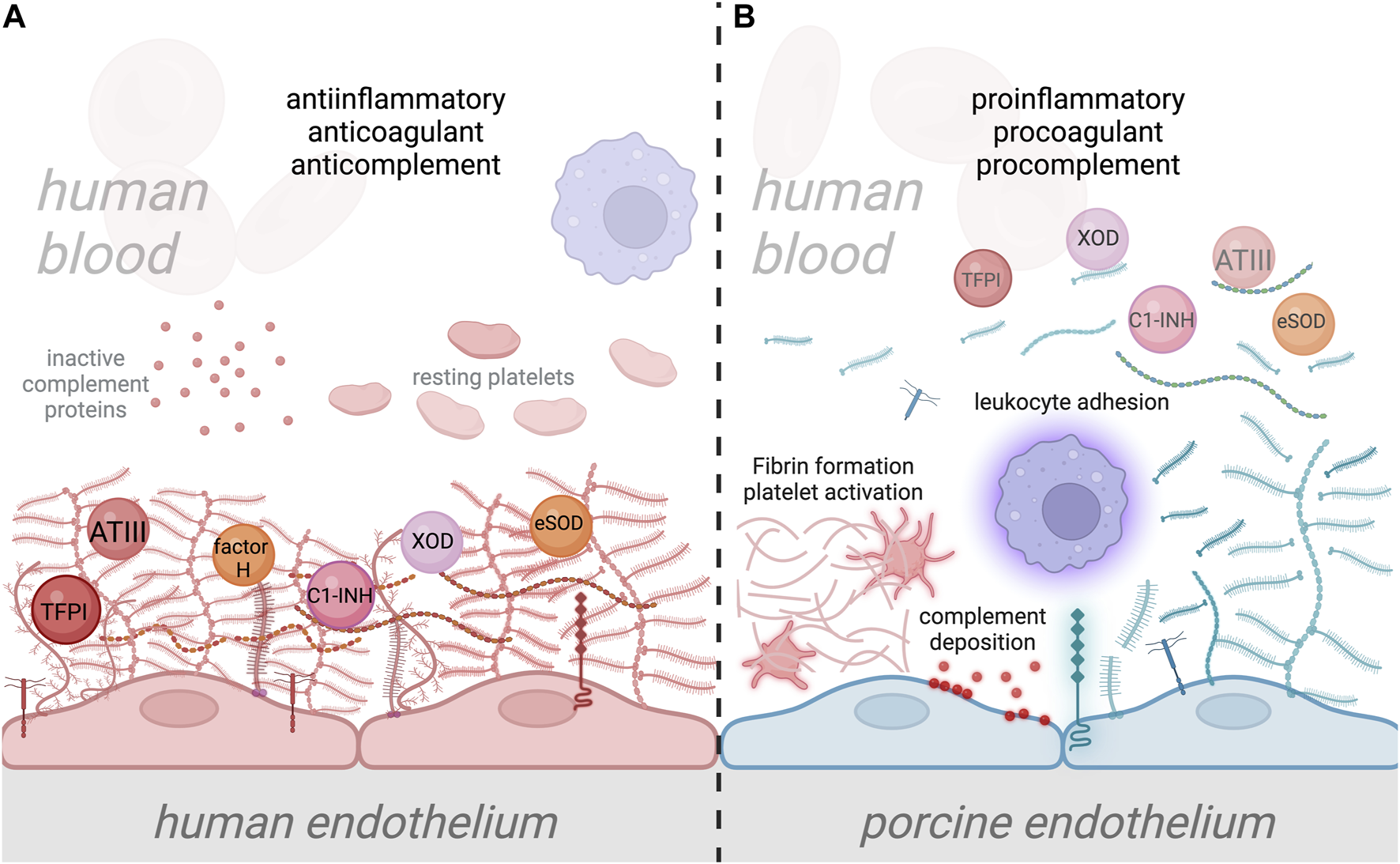
Interaction of human plasma cascade proteins with human and porcine glycocalyx. (A) Normal interaction between human plasma proteins and the human endothelial cell (EC) glycocalyx, where plasma cascade regulatory molecules such as antithrombin III (ATIII), tissue factor pathway inhibitor (TFPI), Factor H, and C1-inhibitor (C1-INH) are specifically bound to EC glycocalyx, providing anticomplement, anticoagulant, and anti-inflammatory properties to the ECs [44, 112–114]. The human EC glycocalyx also binds antioxidant molecules such as XOD and SOD, thereby preventing oxidative damage to ECs [106, 107]. Attachment of leukocytes is prevented by the negatively charged EC glycocalyx and the shielding of leukocyte binding receptors [138, 157]. (B) In xenotransplantation, the disparity between human and porcine EC glycocalyx composition, which likely alters its structure and function, could lead to inefficient binding of protective plasma proteins. This could result in the transition of the endothelium from a quiescent into an activated state, which leads to glycocalyx shedding. Disruption of the porcine EC glycocalyx and loss of its anticoagulant properties will result in activation of the coagulation system, platelet recruitment and fibrin formation. Loss of anticomplement properties will also lead to the deposition of complement proteins on the EC surface, along with the amplification of inflammation. Additionally, shedding of the porcine EC glycocalyx will increase leukocyte attachment to the surface of EC due to the loss of leukocyte binding receptor shielding. Ultimately, this condition could lead to xenograft damage and failure.
Initially, the removal of sugar xenoantigens from xenograft donor pigs by genetic engineering was implemented to overcome the antibody-mediated rejection and has been proven effective [20, 24, 70, 77]. However, these genetic modifications have been found to influence the expression of other sugars and may even lead to the synthesis of novel glycans, which is likely to influence the structure, composition, and immunological function of the porcine EC glycocalyx [159, 160]. Therefore, further studies are necessary to determine how these alterations in sugar composition and the emergence of neoglycans influence the phenotype and physiological function of the porcine EC glycocalyx.
Exploring EC Glycocalyx Protection in Xenotransplantation
Protecting ECs and their glycocalyx is critical for improving graft survival and function, highlighting the importance of targeted therapy for preserving and maintaining the quiescent state of the endothelium. Current strategies, including genetic engineering, might indirectly protect the glycocalyx to a certain extent by preventing antibody binding, complement activation, and controlling the coagulation system. Additionally, anti-inflammatory and anticoagulant agents, such as corticosteroids and heparin, which are already incorporated in current immunosuppressive regimens in xenotransplantation, have been shown to prevent the degradation of the glycocalyx [161–163]. However, although evidence specific to xenotransplantation remains limited, shedding of the glycocalyx continues to be a significant issue in pathological conditions related to vascular inflammation. Shedding of the glycocalyx in a setting of (xeno)transplantation occurs immediately after graft reperfusion (ischemia reperfusion injury), related to trauma and inflammation, or long-term related to chronic rejection [136–138, 156, 164, 165]. Therefore, strategies to prevent shedding of the glycocalyx, or to functionally replace glycocalyx components, might be needed in clinical xenotransplantation.
Generally, glycocalyx protection strategies employ therapeutic agents that interact with one or multiple glycocalyx-modifying factors, including inhibiting the glycocalyx-degrading enzymes, oxidative stress, and inflammation. Molecules such as angiopoietin, hydrocortisone, ATIII, berberine, and S1P prevent the degradation of glycocalyx through the inhibition of MMPs [10, 153, 166, 167]. SOD acts as antioxidant and helps to mitigate oxidative stress associated with ischemia-reperfusion injury, thus protecting the glycocalyx [149]. Sulodexide can protect the glycocalyx by inhibiting heparanase-1 [168]. While those therapies have been used in other vascular disorders, the protective effects on porcine xenografts need further evaluation, especially since existing studies do not fully address the structure, function, and dynamics of the glycocalyx in a xenotransplantation context.
Glycan-specific approaches have also been employed for glycocalyx protection and restoration. Emerging therapies such as bioengineering approaches also offer innovative strategies, for instance, by engineering the EC to rebuild the glycocalyx, which has been shown to minimize graft injury and rejection [169]. Other strategies use a class of molecules which can be termed “endothelial cell protectants.” One such molecule, low molecular weight dextran sulfate of 5000 MW (DXS), was shown to be protective both in vitro and in a small animal model of xenotransplantation [170, 171]. DXS has also been shown to prevent ischemia/reperfusion injury in large animal models [172, 173]. Similarly, multimeric sulfated tyrosine has been shown to help maintain and restore the glycocalyx layer [174]. While currently there are no clinically approved drugs which act as EC protectants, this class of substances has proved to be successful in preclinical experiments and, in our view, by keeping the endothelium in a quiescent state, has great potential as an additional drug treatment in xenotransplantation.
Conclusion
The interaction between the porcine glycocalyx and the human plasma cascades involves complex, yet poorly understood dynamics that are critical to the success of xenotransplantation. To address these challenges, a deeper understanding of interspecies glycocalyx differences and their impact on immune responses, coagulation, and EC function is required. New insights into the structure and function of the porcine and human glycocalyx, as well as the mechanisms of glycocalyx degradation and regeneration in xenotransplantation, hold the potential to unlock novel approaches to preserve the endothelial glycocalyx. Protective strategies for the glycocalyx, combined with the already available strategies to prevent complement and coagulation dysregulation, can be explored as an approach to improving the outcomes of xenotransplantation.
Biography
Robert Rieben was born in Bern, Switzerland, where he also studied biology. During his PhD he worked on blood group ABO antibodies and, via contacts with Rafael Oriol and David Cooper, got in touch with xenotransplantation. As a postdoc in Mohammed Daha’s lab in Leiden/NL, he started to work on the complement system and endothelial cells. At the same time, he collaborated in the first EU-funded xenotransplantation research projects “Glycoimmunology”, led by David Joziasse, and “Xenotransplantation”, led by Bo Samuelsson. Back in Switzerland, he started his own research group and continued to work on the concept of endothelial cell protection in xenotransplantation, allotransplantation, and ischemia/reperfusion injury. For the past 12 years, Robert Rieben has been affiliated to the German Transregio 127 research project on xenotransplantation, led by Bruno Reichart and Eckhard Wolf, and since 2020 he leads a Sinergia project of the Swiss National Science Foundation together with Joerg Seebach and Eckhard Wolf.
Statements
Author contributions
MG: writing (original draft, review, editing). RR: writing (review, editing), supervision. All authors contributed to the article and approved the submitted version.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was funded by the Swiss National Science Foundation grant (SINERGIA Xeno2Cure, grant agreement No. CRSII5198577).
Acknowledgments
The authors would like to thank Jane Shaw (Department for Biomedical Research, University of Bern) for giving helpful suggestions to the manuscript. Figures were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
Ricard N Bailly S Guignabert C Simons M . The Quiescent Endothelium: Signalling Pathways Regulating Organ-Specific Endothelial Normalcy. Nat Rev Cardiol (2021) 18:565–80. 10.1038/s41569-021-00517-4
2.
Lerman A Burnett JC . Intact and Altered Endothelium in Regulation of Vasomotion. Circulation (1992) 86:III12–9.
3.
Li Y-SJ Haga JH Chien S . Molecular Basis of the Effects of Shear Stress on Vascular Endothelial Cells. J Biomech (2005) 38:1949–71. 10.1016/j.jbiomech.2004.09.030
4.
Yau JW Teoh H Verma S . Endothelial Cell Control of Thrombosis. BMC Cardiovasc Disord (2015) 15:130. 10.1186/s12872-015-0124-z
5.
Del Vecchio PJ Siflinger-Birnboim A Shepard JM Bizios R Cooper JA Malik AB . Endothelial Monolayer Permeability to Macromolecules. Fed Proc (1987) 46:2511–5.
6.
Amersfoort J Eelen G Carmeliet P . Immunomodulation by Endothelial Cells — Partnering Up With the Immune System?Nat Rev Immunol (2022) 22:576–88. 10.1038/s41577-022-00694-4
7.
Nieuwdorp M Meuwese MC Vink H Hoekstra JBL Kastelein JJP Stroes ESG . The Endothelial Glycocalyx: A Potential Barrier Between Health and Vascular Disease. Curr Opin Lipidol (2005) 16:507–11. 10.1097/01.mol.0000181325.08926.9c
8.
Luft JH . Fine Structures of Capillary and Endocapillary Layer as Revealed by Ruthenium Red. Fed Proc (1966) 25:1773–83.
9.
Sieve I Münster-Kühnel AK Hilfiker-Kleiner D . Regulation and Function of Endothelial Glycocalyx Layer in Vascular Diseases. Vascul Pharmacol (2018) 100:26–33. 10.1016/j.vph.2017.09.002
10.
Milusev A Rieben R Sorvillo N . The Endothelial Glycocalyx: A Possible Therapeutic Target in Cardiovascular Disorders. Front Cardiovasc Med (2022) 9:897087. 10.3389/fcvm.2022.897087
11.
Banz Y Rieben R . Endothelial Cell Protection in Xenotransplantation: Looking After a Key Player in Rejection. Xenotransplantation (2006) 13:19–30. 10.1111/j.1399-3089.2005.00266.x
12.
Hammer CR . Nature’s Obstacles to Xenotransplantation. Transpl Rev. (1994) 8:174–84. 10.1016/s0955-470x(05)80035-7
13.
Platt JL Bach FH . The Barrier to Xenotransplantation. Transplantation (1991) 52:937–47. 10.1097/00007890-199112000-00001
14.
Cascalho M Platt JL . The Immunological Barrier to Xenotransplantation. Immunity (2001) 14:437–46. 10.1016/s1074-7613(01)00124-8
15.
Stevens RB Platt JL . The Pathogenesis of Hyperacute Xenograft Rejection. Am J Kidney Dis (1992) 20:414–21. 10.1016/s0272-6386(12)70310-4
16.
Lu T Yang B Wang R Qin C . Xenotransplantation: Current Status in Preclinical Research. Front Immunol (2020) 10:3060. 10.3389/fimmu.2019.03060
17.
Gollackner B Goh SK Qawi I Buhler L Knosalla C Daniel S et al Acute Vascular Rejection of Xenografts: Roles of Natural and Elicited Xenoreactive Antibodies in Activation of Vascular Endothelial Cells and Induction of Procoagulant Activity. Transplantation (2004) 77:1735–41. 10.1097/01.tp.0000131167.21930.b8
18.
Shimizu A Meehan SM Kozlowski T Sablinski T Ierino FL Cooper DK et al Acute Humoral Xenograft Rejection: Destruction of the Microvascular Capillary Endothelium in Pig-To-Nonhuman Primate Renal Grafts. Lab Investig J Tech Methods Pathol (2000) 80:815–30. 10.1038/labinvest.3780086
19.
Crikis S Cowan PJ d’Apice AJF . Intravascular Thrombosis in Discordant Xenotransplantation. Transplantation (2006) 82:1119–23. 10.1097/01.tp.0000238721.88920.ee
20.
Phelps CJ Koike C Vaught TD Boone J Wells KD Chen S-H et al Production of α1,3-Galactosyltransferase-Deficient Pigs. Science (2003) 299:411–414. 10.1126/science.1078942
21.
Estrada JL Martens G Li P Adams A Newell KA Ford ML et al Evaluation of Human and Non-Human Primate Antibody Binding to Pig Cells Lacking GGTA1/CMAH/β4GalNT2 Genes. Xenotransplantation (2015) 22:194–202. 10.1111/xen.12161
22.
Zhu A Hurst R . Anti-N-glycolylneuraminic Acid Antibodies Identified in Healthy Human Serum: Anti-NeuGc Antibodies in Human Serum. Xenotransplantation (2002) 9:376–81.
23.
Klymiuk N Aigner B Brem G Wolf E . Genetic Modification of Pigs as Organ Donors for Xenotransplantation. Mol Reprod Dev (2010) 77:209–21. 10.1002/mrd.21127
24.
Cooper DKC Hara H Iwase H Yamamoto T Li Q Ezzelarab M et al Justification of Specific Genetic Modifications in Pigs for Clinical Organ Xenotransplantation. Xenotransplantation (2019) 26:e12516. 10.1111/xen.12516
25.
Ali A Kemter E Wolf E . Advances in Organ and Tissue Xenotransplantation. Annu Rev Anim Biosci (2024) 12:369–90. 10.1146/annurev-animal-021122-102606
26.
Griffith BP Goerlich CE Singh AK Rothblatt M Lau CL Shah A et al Genetically Modified Porcine-To-Human Cardiac Xenotransplantation. N Engl J Med (2022) 387:35–44. 10.1056/NEJMoa2201422
27.
Porrett PM Orandi BJ Kumar V Houp J Anderson D Cozette Killian A et al First Clinical-Grade Porcine Kidney Xenotransplant Using a Human Decedent Model. Am J Transpl (2022) 22:1037–53. 10.1111/ajt.16930
28.
Ekser B Long C Echeverri GJ Hara H Ezzelarab M Lin CC et al Impact of Thrombocytopenia on Survival of Baboons with Genetically Modified Pig Liver Transplants: Clinical Relevance. Am J Transpl (2010) 10:273–85. 10.1111/j.1600-6143.2009.02945.x
29.
Chaban R Cooper DKC Pierson RN . Pig Heart and Lung Xenotransplantation: Present Status. J Heart Lung Transpl (2022) 41:1014–22. 10.1016/j.healun.2022.04.010
30.
Iwase H Hara H Ezzelarab M Li T Zhang Z Gao B et al Immunological and Physiological Observations in Baboons with Life‐Supporting Genetically Engineered Pig Kidney Grafts. Xenotransplantation (2017) 24:e12293. 10.1111/xen.12293
31.
Longstaff C Kolev K . Basic Mechanisms and Regulation of Fibrinolysis. J Thromb Haemost (2015) 13:S98–S105. 10.1111/jth.12935
32.
Maas C Renné T . Coagulation Factor XII in Thrombosis and Inflammation. Blood (2018) 131:1903–9. 10.1182/blood-2017-04-569111
33.
Kleinschnitz C Stoll G Bendszus M Schuh K Pauer HU Burfeind P et al Targeting Coagulation Factor XII Provides Protection From Pathological Thrombosis in Cerebral Ischemia Without Interfering With Hemostasis. J Exp Med (2006) 203:513–8. 10.1084/jem.20052458
34.
Neubauer K Zieger B . Endothelial Cells and Coagulation. Cell Tissue Res. (2022) 387:391–8. 10.1007/s00441-021-03471-2
35.
Moncada S Higgs EA Vane JR . Human Arterial and Venous Tissues Generate Prostacyclin (Prostaglandin X), a Potent Inhibitor of Platelet Aggregation. Lancet Lond Engl (1977) 1:18–20. 10.1016/s0140-6736(77)91655-5
36.
Radomski MW Palmer RMJ Moncada S . Endogenous Nitric Oxide Inhibits Human Platelet Adhesion to Vascular Endothelium. Lancet (1987) 330:1057–8. 10.1016/s0140-6736(87)91481-4
37.
Radomski MW Palmer RM Moncada S . An L-Arginine/Nitric Oxide Pathway Present in Human Platelets Regulates Aggregation. Proc Natl Acad Sci U S A (1990) 87:5193–7. 10.1073/pnas.87.13.5193
38.
Maroney SA Mast AE . Expression of Tissue Factor Pathway Inhibitor by Endothelial Cells and Platelets. Transfus Apher Sci (2008) 38:9–14. 10.1016/j.transci.2007.12.001
39.
Ameri A Kuppuswamy M Basu S Bajaj S . Expression of Tissue Factor Pathway Inhibitor by Cultured Endothelial Cells in Response to Inflammatory Mediators. Blood (1992) 79:3219–26. 10.1182/blood.v79.12.3219.bloodjournal79123219
40.
Wood JP Bunce MW Maroney SA Tracy PB Camire RM Mast AE . Tissue Factor Pathway Inhibitor-Alpha Inhibits Prothrombinase During the Initiation of Blood Coagulation. Proc Natl Acad Sci (2013) 110:17838–43. 10.1073/pnas.1310444110
41.
Loskutoff DJ Edgington TE . Synthesis of a Fibrinolytic Activator and Inhibitor by Endothelial Cells. Proc Natl Acad Sci U S A (1977) 74:3903–7. 10.1073/pnas.74.9.3903
42.
Jaffe EA . Cell Biology of Endothelial Cells. Hum Pathol (1987) 18:234–9. 10.1016/s0046-8177(87)80005-9
43.
Bitomsky W Wade RC . Docking of Glycosaminoglycans to Heparin-Binding Proteins: Validation for aFGF, bFGF, and Antithrombin and Application to IL-8. J Am Chem Soc (1999) 121:3004–13. 10.1021/ja983319g
44.
De Agostini AI Watkins SC Slayter HS Youssoufian H Rosenberg RD . Localization of Anticoagulantly Active Heparan Sulfate Proteoglycans in Vascular Endothelium: Antithrombin Binding on Cultured Endothelial Cells and Perfused Rat Aorta. J Cel Biol. (1990) 111:1293–304. 10.1083/jcb.111.3.1293
45.
Bauer KA Rosenberg RD . Role of Antithrombin III as a Regulator of In Vivo Coagulation. Semin Hematol (1991) 28:10–8.
46.
Hofsteenge J Taguchi H Stone SR . Effect of Thrombomodulin on the Kinetics of the Interaction of Thrombin With Substrates and Inhibitors. Biochem J (1986) 237:243–51. 10.1042/bj2370243
47.
Esmon N Esmon C . Protein C and the Endothelium. Semin Thromb Hemost (1988) 14:210–5. 10.1055/s-2007-1002779
48.
Stearns-Kurosawa DJ Kurosawa S Mollica JS Ferrell GL Esmon CT . The Endothelial Cell Protein C Receptor Augments Protein C Activation by the Thrombin-Thrombomodulin Complex. Proc Natl Acad Sci (1996) 93:10212–6. 10.1073/pnas.93.19.10212
49.
Gulati P Lemercier C Guc D Lappin D Whaley K . Regulation of the Synthesis of C1 Subcomponents and C1-Inhibitor. Behring Inst Mitt (1993) 196–203:196–203.
50.
McNearney T Ballard L Seya T Atkinson JP . Membrane Cofactor Protein of Complement Is Present on Human Fibroblast, Epithelial, and Endothelial Cells. J Clin Invest (1989) 84:538–45. 10.1172/JCI114196
51.
Asch AS Kinoshita T Jaffe EA Nussenzweig V . Decay-Accelerating Factor Is Present on Cultured Human Umbilical Vein Endothelial Cells. J Exp Med (1986) 163:221–6. 10.1084/jem.163.1.221
52.
Brooimans RA Hiemstra PS van der Ark AA Sim RB van Es LA Daha MR . Biosynthesis of Complement Factor H by Human Umbilical Vein Endothelial Cells. Regulation by T Cell Growth Factor and IFN-Gamma. J Immunol Baltim Md 1950 (1989) 142:2024–30. 10.4049/jimmunol.142.6.2024
53.
Julen N Dauchel H Lemercier C Sim RB Fontaine M Ripoche J . In vitro Biosynthesis of Complement Factor I by Human Endothelial Cells. Eur J Immunol (1992) 22:213–7. 10.1002/eji.1830220131
54.
Hamilton K Ji Z Rollins S Stewart B Sims P . Regulatory Control of the Terminal Complement Proteins at the Surface of Human Endothelial Cells: Neutralization of a C5b-9 Inhibitor by Antibody to CD59. Blood (1990) 76:2572–7. 10.1182/blood.v76.12.2572.bloodjournal76122572
55.
Berge V Johnson E Høgåsen K . Clusterin and the Terminal Complement Pathway Synthesized by Human Umbilical Vein Endothelial Cells Are Closely Linked When Detected on Co‐Cultured Agarose Beads. APMIS (1997) 105:17–24. 10.1111/j.1699-0463.1997.tb00534.x
56.
Urbich C Fritzenwanger M Zeiher AM Dimmeler S . Laminar Shear Stress Upregulates the Complement-Inhibitory Protein Clusterin: A Novel Potent Defense Mechanism Against Complement-Induced Endothelial Cell Activation. Circulation (2000) 101:352–5. 10.1161/01.cir.101.4.352
57.
Nonaka M . Evolution of the Complement System. Curr Opin Immunol (2001) 13:69–73. 10.1016/s0952-7915(00)00184-9
58.
Dzik S . Complement and Coagulation: Cross Talk Through Time. Transfus Med Rev (2019) 33:199–206. 10.1016/j.tmrv.2019.08.004
59.
Ghebrehiwet B Silverberg M Kaplan AP . Activation of the Classical Pathway of Complement by Hageman Factor Fragment. J Exp Med (1981) 153:665–76. 10.1084/jem.153.3.665
60.
Foley JH Walton BL Aleman MM O'Byrne AM Lei V Harrasser M et al Complement Activation in Arterial and Venous Thrombosis Is Mediated by Plasmin. EBioMedicine (2016) 5:175–82. 10.1016/j.ebiom.2016.02.011
61.
Mahdi F Madar ZS Figueroa CD Schmaier AH . Factor XII Interacts With the Multiprotein Assembly of Urokinase Plasminogen Activator Receptor, gC1qR, and Cytokeratin 1 on Endothelial Cell Membranes. Blood (2002) 99:3585–96. 10.1182/blood.v99.10.3585
62.
Amara U Rittirsch D Flierl M Bruckner U Klos A Gebhard F et al Interaction Between the Coagulation and Complement System. in Current Topics in Complement II ed. LambrisJD (New York, NY: Springer US) (2008) vol. 63271–9.
63.
Conway EM . Complement-Coagulation Connections. Blood Coagul Fibrinolysis (2018) 29:243–51. 10.1097/MBC.0000000000000720
64.
Cugno M Cicardi M Bottasso B Coppola R Paonessa R Mannucci PM et al Activation of the Coagulation Cascade in C1-Inhibitor Deficiencies. Blood (1997) 89:3213–8. 10.1182/blood.V89.9.3213
65.
Oikonomopoulou K Ricklin D Ward PA Lambris JD . Interactions Between Coagulation and Complement—Their Role in Inflammation. Semin Immunopathol (2012) 34:151–65. 10.1007/s00281-011-0280-x
66.
Delvaeye M Noris M De Vriese A Esmon CT Esmon NL Ferrell G et al Thrombomodulin Mutations in Atypical Hemolytic–Uremic Syndrome. N Engl J Med (2009) 361:345–57. 10.1056/NEJMoa0810739
67.
Kluge KE Langseth MS Bratseth V Pettersen AÅ Arnesen H Tønnessen T et al Circulating Levels of the Terminal Complement Complex Are Associated With Hypercoagulability in Patients With Stable Coronary Artery Disease. Thromb Res (2020) 196:106–8. 10.1016/j.thromres.2020.08.023
68.
Irie A Koyama S Kozutsumi Y Kawasaki T Suzuki A . The Molecular Basis for the Absence of N-Glycolylneuraminic Acid in Humans. J Biol Chem (1998) 273:15866–71. 10.1074/jbc.273.25.15866
69.
Platt JL Lindman BJ Geller RL Noreen HJ Swanson JL Dalmasso AP et al The Role of Natural Antibodies in the Activation of Xenogenic Endothelial Cells. Transplantation (1991) 52:1037–43. 10.1097/00007890-199112000-00019
70.
Chen G Qian H Starzl T Sun H Garcia B Wang X et al Acute Rejection Is Associated With Antibodies to Non-Gal Antigens in Baboons Using Gal-Knockout Pig Kidneys. Nat Med (2005) 11:1295–8. 10.1038/nm1330
71.
Cooper DK Good AH Koren E Oriol R Malcolm AJ Ippolito RM et al Identification of Alpha-Galactosyl and Other Carbohydrate Epitopes That Are Bound by Human Anti-Pig Antibodies: Relevance to Discordant Xenografting in Man. Transpl Immunol (1993) 1:198–205. 10.1016/0966-3274(93)90047-c
72.
Varki NM Varki A . Diversity in Cell Surface Sialic Acid Presentations: Implications for Biology and Disease. Lab Invest (2007) 87:851–7. 10.1038/labinvest.3700656
73.
Byrne GW Stalboerger PG Du Z Davis TR McGregor CGA . Identification of New Carbohydrate and Membrane Protein Antigens in Cardiac Xenotransplantation. Transplantation (2011) 91:287–92. 10.1097/TP.0b013e318203c27d
74.
Cowan PJ Robson SC dʼApice AJ . Controlling Coagulation Dysregulation in Xenotransplantation. Curr Opin Organ Transpl (2011) 16:214–21. 10.1097/MOT.0b013e3283446c65
75.
Lam TT Paniagua R Shivaram G Schuurman HJ Borie DC Morris RE . Anti‐Non‐Gal Porcine Endothelial Cell Antibodies in Acute Humoral Xenograft Rejection of hDAF‐Transgenic Porcine Hearts in Cynomolgus Monkeys. Xenotransplantation (2004) 11:531–5. 10.1111/j.1399-3089.2004.00175.x
76.
Cooper DKC Ezzelarab MB Hara H Iwase H Lee W Wijkstrom M et al The Pathobiology of Pig‐To‐Primate Xenotransplantation: A Historical Review. Xenotransplantation (2016) 23:83–105. 10.1111/xen.12219
77.
Lutz AJ Li P Estrada JL Sidner RA Chihara RK Downey SM et al Double Knockout Pigs Deficient in N ‐Glycolylneuraminic Acid and G Alactose α‐1,3‐ G Alactose Reduce the Humoral Barrier to Xenotransplantation. Xenotransplantation (2013) 20:27–35. 10.1111/xen.12019
78.
Adams DH Kadner A Chen RH Farivar RS . Human Membrane Cofactor Protein (MCP, CD 46) Protects Transgenic Pig Hearts From Hyperacute Rejection in Primates: CD 46 Protein. Xenotransplantation (2001) 8:36–40. 10.1046/j.0908-665x.2000.00085.x
79.
Chen RH Naficy S Logan JS Diamond LE Adams DH . Hearts From Transgenic Pigs Constructed With CD59/DAF Genomic Clones Demonstrate Improved Survival in Primates: Improved Primate Survival With Transgenic Pig Hearts. Xenotransplantation (1999) 6:194–200. 10.1034/j.1399-3089.1999.00017.x
80.
Lee KFE Salvaris EJ Roussel JC Robson SC d'Apice AJF Cowan PJ . Recombinant Pig TFPI Efficiently Regulates Human Tissue Factor Pathways. Xenotransplantation (2008) 15:191–7. 10.1111/j.1399-3089.2008.00476.x
81.
Längin M Mayr T Reichart B Michel S Buchholz S Guethoff S et al Consistent Success in Life-Supporting Porcine Cardiac Xenotransplantation. Nature (2018) 564:430–3. 10.1038/s41586-018-0765-z
82.
Wheeler DG Joseph ME Mahamud SD Aurand WL Mohler PJ Pompili VJ et al Transgenic Swine: Expression of Human CD39 Protects Against Myocardial Injury. J Mol Cel Cardiol. (2012) 52:958–61. 10.1016/j.yjmcc.2012.01.002
83.
Lee S-C Lee H Oh KB Hwang IS Yang H Park MR et al Production and Breeding of Transgenic Cloned Pigs Expressing Human CD73. Dev Reprod (2017) 21:157–65. 10.12717/DR.2017.21.2.157
84.
Connolly MR Kuravi K Burdorf L Sorrells L Morrill B Cimeno A et al Humanized Von Willebrand Factor Reduces Platelet Sequestration in Ex Vivo and In Vivo Xenotransplant Models. Xenotransplantation (2021) 28:e12712. 10.1111/xen.12712
85.
Paris LL Chihara RK Reyes LM Sidner RA Estrada JL Downey SM et al ASGR1 Expressed by Porcine Enriched Liver Sinusoidal Endothelial Cells Mediates Human Platelet Phagocytosis In Vitro. Xenotransplantation (2011) 18:245–51. 10.1111/j.1399-3089.2011.00639.x
86.
Paris LL Estrada JL Li P Blankenship RL Sidner RA Reyes LM et al Reduced Human Platelet Uptake by Pig Livers Deficient in the Asialoglycoprotein Receptor 1 Protein. Xenotransplantation (2015) 22:203–10. 10.1111/xen.12164
87.
Mohiuddin MM Singh AK Corcoran PC Thomas Iii ML Clark T Lewis BG et al Chimeric 2C10R4 Anti-CD40 Antibody Therapy Is Critical for Long-Term Survival of GTKO.hCD46.hTBM Pig-To-Primate Cardiac Xenograft. Nat Commun (2016) 7:11138. 10.1038/ncomms11138
88.
Kim SC Mathews DV Breeden CP Higginbotham LB Ladowski J Martens G et al Long-Term Survival of Pig-To-Rhesus Macaque Renal Xenografts Is Dependent on CD4 T Cell Depletion. Am J Transpl (2019) 19:2174–85. 10.1111/ajt.15329
89.
Montgomery RA Stern JM Lonze BE Tatapudi VS Mangiola M Wu M et al Results of Two Cases of Pig-To-Human Kidney Xenotransplantation. N Engl J Med (2022) 386:1889–98. 10.1056/NEJMoa2120238
90.
Fu BM Tarbell JM . Mechano‐Sensing and Transduction by Endothelial Surface Glycocalyx: Composition, Structure, and Function. Wires Syst Biol Med (2013) 5:381–90. 10.1002/wsbm.1211
91.
Ebong EE Lopez-Quintero SV Rizzo V Spray DC Tarbell JM . Shear-Induced Endothelial NOS Activation and Remodeling via Heparan Sulfate, Glypican-1, and Syndecan-1. Integr Biol (2014) 6:338–47. 10.1039/c3ib40199e
92.
Wandall HH Rumjantseva V Sørensen ALT Patel-Hett S Josefsson EC Bennett EP et al The Origin and Function of Platelet Glycosyltransferases. Blood (2012) 120:626–35. 10.1182/blood-2012-02-409235
93.
Manhardt CT Punch PR Dougher CWL Lau JTY . Extrinsic Sialylation Is Dynamically Regulated by Systemic Triggers In Vivo. J Biol Chem (2017) 292:13514–20. 10.1074/jbc.C117.795138
94.
Varki A . Essentials of Glycobiology. Long Island, New York: Cold Spring Harbor Laboratory Press, 2015.
95.
Antonelli A D’Amore PA . Density-Dependent Expression of Hyaluronic Acid Binding to Vascular Cells In Vitro. Microvasc Res (1991) 41:239–51. 10.1016/0026-2862(91)90025-7
96.
Merrilees MJ Scott L . Culture of Rat and Pig Aortic Endothelial Cells Differences in Their Isolation, Growth Rate and Glycosaminoglycan Synthesis. Atherosclerosis (1981) 38:19–26. 10.1016/0021-9150(81)90099-x
97.
Ribeiro MMB Domingues MM Freire JM Santos NC Castanho MARB . Translocating the Blood-Brain Barrier Using Electrostatics. Front Cel Neurosci. (2012) 6:44. 10.3389/fncel.2012.00044
98.
Walter FR Santa-Maria AR Mészáros M Veszelka S Dér A Deli MA . Surface Charge, Glycocalyx, and Blood-Brain Barrier Function. Tissue Barriers (2021) 9:1904773. 10.1080/21688370.2021.1904773
99.
Rubanyi GM Romero JC Vanhoutte PM . Flow-Induced Release of Endothelium-Derived Relaxing Factor. Am J Physiol.-Heart Circ Physiol (1986) 250:H1145–9. 10.1152/ajpheart.1986.250.6.H1145
100.
Dardik R Lahav J . The Structure of Endothelial Cell Thrombospondin: Characterization of the Heparin‐Binding Domains. Eur J Biochem (1987) 168:347–55. 10.1111/j.1432-1033.1987.tb13426.x
101.
Karumanchi SA Jha V Ramchandran R Karihaloo A Tsiokas L Chan B et al Cell Surface Glypicans Are Low-Affinity Endostatin Receptors. Mol Cel (2001) 7:811–22. 10.1016/s1097-2765(01)00225-8
102.
Rein CM Desai UR Church FC . Serpin–Glycosaminoglycan Interactions. Methods Enzymol (2011) 501:105–37. Elsevier. 10.1016/B978-0-12-385950-1.00007-9
103.
DiGabriele AD Lax I Chen DI Svahn CM Jaye M Schlessinger J et al Structure of a Heparin-Linked Biologically Active Dimer of Fibroblast Growth Factor. Nature (1998) 393:812–7. 10.1038/31741
104.
Rossi D Zlotnik A . The Biology of Chemokines and Their Receptors. Annu Rev Immunol (2000) 18:217–42. 10.1146/annurev.immunol.18.1.217
105.
Van Wijk XMR Van Kuppevelt TH . Heparan Sulfate in Angiogenesis: A Target for Therapy. Angiogenesis (2013) 17:443–62. 10.1007/s10456-013-9401-6
106.
Adachi T Fukushima T Usami Y Hirano K . Binding of Human Xanthine Oxidase to Sulphated Glycosaminoglycans on the Endothelial-Cell Surface. Biochem J (1993) 289:523–7. 10.1042/bj2890523
107.
Becker M Menger MD Lehr HA . Heparin-Released Superoxide Dismutase Inhibits Postischemic Leukocyte Adhesion to Venular Endothelium. Am J Physiol.-Heart Circ Physiol (1994) 267:H925–30. 10.1152/ajpheart.1994.267.3.H925
108.
Rosenberg RD Shworak NW Liu J Schwartz JJ Zhang L . Heparan Sulfate Proteoglycans of the Cardiovascular System. Specific Structures Emerge But How Is Synthesis Regulated?J Clin Invest (1997) 99:2062–70. 10.1172/JCI119377
109.
Sarrazin S Lamanna WC Esko JD . Heparan Sulfate Proteoglycans. Cold Spring Harb Perspect Biol (2011) 3:a004952. 10.1101/cshperspect.a004952
110.
De Mattos DA Stelling MP Tovar AMF Mourão PAS . Heparan Sulfates From Arteries and Veins Differ in Their Antithrombin-Mediated Anticoagulant Activity. J Thromb Haemost (2008) 6:1987–90. 10.1111/j.1538-7836.2008.03145.x
111.
Borza D-B . Glomerular Basement Membrane Heparan Sulfate in Health and Disease: A Regulator of Local Complement Activation. Matrix Biol (2017) 57–58:299–310. 10.1016/j.matbio.2016.09.002
112.
Wuillemin WA te Velthuis H Lubbers YT de Ruig CP Eldering E Hack CE . Potentiation of C1 Inhibitor by Glycosaminoglycans: Dextran Sulfate Species Are Effective Inhibitors of In Vitro Complement Activation in Plasma. J Immunol (1997) 159:1953–60. 10.4049/jimmunol.159.4.1953
113.
Perkins SJ Fung KW Khan S . Molecular Interactions Between Complement Factor H and Its Heparin and Heparan Sulfate Ligands. Front Immunol (2014) 5:126. 10.3389/fimmu.2014.00126
114.
Colburn P Buonassisi V . Anti-Clotting Activity of Endothelial Cell Cultures and Heparan Sulfate Proteoglycans. Biochem Biophys Res Commun (1982) 104:220–7. 10.1016/0006-291x(82)91962-3
115.
Wiedermann CJ Römisch J . The Anti‐Inflammatory Actions of Antithrombin – A Review. Acta Med Austriaca (2002) 29:89–92. 10.1046/j.1563-2571.2002.02012.x
116.
Kassam G Manro A Braat CE Louie P Fitzpatrick SL Waisman DM . Characterization of the Heparin Binding Properties of Annexin II Tetramer. J Biol Chem (1997) 272:15093–100. 10.1074/jbc.272.24.15093
117.
Yan Z Liu J Xie L Liu X Zeng Y . Role of Heparan Sulfate in Mediating CXCL8-Induced Endothelial Cell Migration. PeerJ (2016) 4:e1669. 10.7717/peerj.1669
118.
Norgard-Sumnicht K Varki A . Endothelial Heparan Sulfate Proteoglycans That Bind to L-Selectin Have Glucosamine Residues With Unsubstituted Amino Groups. J Biol Chem (1995) 270:12012–24. 10.1074/jbc.270.20.12012
119.
Nelson R Cecconi O Roberts WG Aruffo A Linhardt RJ Bevilacqua MP . Heparin Oligosaccharides Bind L- and P-Selectin and Inhibit Acute Inflammation. Blood (1993) 82:3253–8. 10.1182/blood.V82.11.3253.3253
120.
Renné T Dedio J David G Müller-Esterl W . High Molecular Weight Kininogen Utilizes Heparan Sulfate Proteoglycans for Accumulation on Endothelial Cells. J Biol Chem (2000) 275:33688–96. 10.1074/jbc.M000313200
121.
Motta G Tersariol ILS . Modulation of the Plasma Kallikrein-Kinin System Proteins Performed by Heparan Sulfate Proteoglycans. Front Physiol (2017) 8:481. 10.3389/fphys.2017.00481
122.
Schauer R . Sialic Acids as Regulators of Molecular and Cellular Interactions. Curr Opin Struct Biol (2009) 19:507–14. 10.1016/j.sbi.2009.06.003
123.
Varki A Gagneux P . Multifarious Roles of Sialic Acids in Immunity. Ann N Y Acad Sci (2012) 1253:16–36. 10.1111/j.1749-6632.2012.06517.x
124.
Varki A . Glycan-Based Interactions Involving Vertebrate Sialic-Acid-Recognizing Proteins. Nature (2007) 446:1023–9. 10.1038/nature05816
125.
Reily C Stewart TJ Renfrow MB Novak J . Glycosylation in Health and Disease. Nat Rev Nephrol (2019) 15:346–66. 10.1038/s41581-019-0129-4
126.
Lasky LA . Selectin-Carbohydrate Interactions and the Initiation of the Inflammatory Response. Annu Rev Biochem (1995) 64:113–39. 10.1146/annurev.bi.64.070195.000553
127.
Zhang Y Zheng Y Li J Nie L Hu Y Wang F et al Immunoregulatory Siglec Ligands Are Abundant in Human and Mouse Aorta and Are Up-Regulated by High Glucose. Life Sci (2019) 216:189–99. 10.1016/j.lfs.2018.11.049
128.
Meri S Pangburn MK . Discrimination Between Activators and Nonactivators of the Alternative Pathway of Complement: Regulation via a Sialic Acid/polyanion Binding Site on Factor H. Proc Natl Acad Sci (1990) 87:3982–6. 10.1073/pnas.87.10.3982
129.
Meri S Pangburn MK . Regulation of Alternative Pathway Complement Activation by Glycosaminoglycans: Specificity of the Polyanion Binding Site on Factor H. Biochem Biophys Res Commun (1994) 198:52–9. 10.1006/bbrc.1994.1008
130.
Abeln M Albers I Peters-Bernard U Flächsig-Schulz K Kats E Kispert A et al Sialic Acid Is a Critical Fetal Defense Against Maternal Complement Attack. J Clin Invest (2018) 129:422–36. 10.1172/JCI99945
131.
Blaum BS Hannan JP Herbert AP Kavanagh D Uhrín D Stehle T . Structural Basis for Sialic Acid–Mediated Self-Recognition by Complement Factor H. Nat Chem Biol (2015) 11:77–82. 10.1038/nchembio.1696
132.
Furukawa K Ohmi Y Ohkawa Y Tokuda N Kondo Y Tajima O et al Regulatory Mechanisms of Nervous Systems With Glycosphingolipids. Neurochem Res (2011) 36:1578–86. 10.1007/s11064-011-0494-2
133.
Furukawa K Ohmi Y Kondo Y Ohkawa Y Tajima O . Regulatory Function of Glycosphingolipids in the Inflammation and Degeneration. Arch Biochem Biophys (2015) 571:58–65. 10.1016/j.abb.2015.02.007
134.
Okuda T Nakakita S Nakayama K . Structural Characterization and Dynamics of Globotetraosylceramide in Vascular Endothelial Cells Under TNF-α Stimulation. Glycoconj J (2010) 27:287–96. 10.1007/s10719-009-9277-2
135.
Kinoshita T Fujita M Maeda Y . Biosynthesis, Remodelling and Functions of Mammalian GPI-Anchored Proteins: Recent Progress. J Biochem (Tokyo) (2008) 144:287–94. 10.1093/jb/mvn090
136.
Zuurbier CJ Demirci C Koeman A Vink H Ince C . Short-Term Hyperglycemia Increases Endothelial Glycocalyx Permeability and Acutely Decreases Lineal Density of Capillaries With Flowing Red Blood Cells. J Appl Physiol (2005) 99:1471–6. 10.1152/japplphysiol.00436.2005
137.
Kozar RA Peng Z Zhang R Holcomb JB Pati S Park P et al Plasma Restoration of Endothelial Glycocalyx in a Rodent Model of Hemorrhagic Shock. Anesth Analg (2011) 112:1289–95. 10.1213/ANE.0b013e318210385c
138.
Mulivor AW Lipowsky HH . Inflammation- and Ischemia-Induced Shedding of Venular Glycocalyx. Am J Physiol.-Heart Circ Physiol (2004) 286:H1672–80. 10.1152/ajpheart.00832.2003
139.
Chignalia AZ Yetimakman F Christiaans SC Unal S Bayrakci B Wagener BM et al The Glycocalyx and Trauma: A Review. Shock (2016) 45:338–48. 10.1097/SHK.0000000000000513
140.
Milusev A Despont A Shaw J Rieben R Sorvillo N . Inflammatory Stimuli Induce Shedding of Heparan Sulfate From Arterial But Not Venous Porcine Endothelial Cells Leading to Differential Proinflammatory and Procoagulant Responses. Sci Rep (2023) 13:4483. 10.1038/s41598-023-31396-z
141.
Platt JL Vercellotti GM Lindman BJ Oegema TR Bach FH Dalmasso AP . Release of Heparan Sulfate From Endothelial Cells. Implications for Pathogenesis of Hyperacute Rejection. J Exp Med (1990) 171:1363–8. 10.1084/jem.171.4.1363
142.
Platt JL Dalmasso AP Lindman BJ Ihrcke NS Bach FH . The Role of C5a and Antibody in the Release of Heparan Sulfate From Endothelial Cells. Eur J Immunol (1991) 21:2887–90. 10.1002/eji.1830211135
143.
Platt JL Wrenshall LE Johnson GB Cascalho M Heparan Sulfate Proteoglycan Metabolism and the Fate of Grafted Tissues. in Immune Responses to Biosurfaces eds. LambrisJDEkdahlKNRicklinDNilssonB (Springer International Publishing, Cham) (2015). vol. 865123–40.
144.
Milusev A Ren J Despont A Shaw J Längin M Bender M et al Glycocalyx Dynamics and the Inflammatory Response of Genetically Modified Porcine Endothelial Cells. Xenotransplantation (2023) 30:e12820. 10.1111/xen.12820
145.
Reitsma S Slaaf DW Vink H van Zandvoort MAMJ oude Egbrink MGA . The Endothelial Glycocalyx: Composition, Functions, and Visualization. Pflugers Arch (2007) 454:345–59. 10.1007/s00424-007-0212-8
146.
Wang W . Change in Properties of the Glycocalyx Affects the Shear Rate and Stress Distribution on Endothelial Cells. J Biomech Eng (2007) 129:324–9. 10.1115/1.2720909
147.
Davies PF . Hemodynamic Shear Stress and the Endothelium in Cardiovascular Pathophysiology. Nat Clin Pract Cardiovasc Med (2009) 6:16–26. 10.1038/ncpcardio1397
148.
Chappell D Jacob M . Role of the Glycocalyx in Fluid Management: Small Things Matter. Best Pract Res Clin Anaesthesiol (2014) 28:227–34. 10.1016/j.bpa.2014.06.003
149.
Rubio-Gayosso I Platts SH Duling BR . Reactive Oxygen Species Mediate Modification of Glycocalyx During Ischemia-Reperfusion Injury. Am J Physiol.-Heart Circ Physiol (2006) 290:H2247–56. 10.1152/ajpheart.00796.2005
150.
Cancel LM Ebong EE Mensah S Hirschberg C Tarbell JM . Endothelial Glycocalyx, Apoptosis and Inflammation in an Atherosclerotic Mouse Model. Atherosclerosis (2016) 252:136–46. 10.1016/j.atherosclerosis.2016.07.930
151.
Van Den Hoven MJ Rops AL Vlodavsky I Levidiotis V Berden JH van der Vlag J . Heparanase in Glomerular Diseases. Kidney Int (2007) 72:543–8. 10.1038/sj.ki.5002337
152.
Subramanian SV Fitzgerald ML Bernfield M . Regulated Shedding of Syndecan-1 and -4 Ectodomains by Thrombin and Growth Factor Receptor Activation. J Biol Chem (1997) 272:14713–20. 10.1074/jbc.272.23.14713
153.
Becker BF Jacob M Leipert S Salmon AHJ Chappell D . Degradation of the Endothelial Glycocalyx in Clinical Settings: Searching for the Sheddases. Br J Clin Pharmacol (2015) 80:389–402. 10.1111/bcp.12629
154.
Abassi Z Armaly Z Heyman SN . Glycocalyx Degradation in Ischemia-Reperfusion Injury. Am J Pathol (2020) 190:752–67. 10.1016/j.ajpath.2019.08.019
155.
Smart L Macdonald SPJ Burrows S Bosio E Arendts G Fatovich DM . Endothelial Glycocalyx Biomarkers Increase in Patients With Infection During Emergency Department Treatment. J Crit Care (2017) 42:304–9. 10.1016/j.jcrc.2017.07.001
156.
Goligorsky MS Sun D . Glycocalyx in Endotoxemia and Sepsis. Am J Pathol (2020) 190:791–8. 10.1016/j.ajpath.2019.06.017
157.
Constantinescu AA Vink H Spaan JAE . Endothelial Cell Glycocalyx Modulates Immobilization of Leukocytes at the Endothelial Surface. Arterioscler Thromb Vasc Biol (2003) 23:1541–7. 10.1161/01.ATV.0000085630.24353.3D
158.
Kuschert GSV Coulin F Power CA Proudfoot AE Hubbard RE Hoogewerf AJ et al Glycosaminoglycans Interact Selectively With Chemokines and Modulate Receptor Binding and Cellular Responses. Biochemistry (1999) 38:12959–68. 10.1021/bi990711d
159.
Burlak C Bern M Brito AE Isailovic D Wang ZY Estrada JL et al N ‐linked Glycan Profiling of GGTA 1/CMAH Knockout Pigs Identifies New Potential Carbohydrate Xenoantigens. Xenotransplantation (2013) 20:277–91. 10.1111/xen.12047
160.
Morticelli L Rossdam C Cajic S Böthig D Magdei M Tuladhar SR et al Genetic Knockout of Porcine GGTA1 or CMAH/GGTA1 Is Associated With the Emergence of Neo‐Glycans. Xenotransplantation (2023) 30:e12804. 10.1111/xen.12804
161.
Johannes T Mik EG Klingel K Dieterich HJ Unertl KE Ince C . Low-Dose Dexamethasone-Supplemented Fluid Resuscitation Reverses Endotoxin-Induced Acute Renal Failure and Prevents Cortical Microvascular Hypoxia. Shock (2009) 31:521–8. 10.1097/SHK.0b013e318188d198
162.
Yini S Heng Z Xin A Xiaochun M . Effect of Unfractionated Heparin on Endothelial Glycocalyx in a Septic Shock Model. Acta Anaesthesiol Scand (2015) 59:160–9. 10.1111/aas.12418
163.
Yang R Chen M Zheng J Li X Zhang X . The Role of Heparin and Glycocalyx in Blood–Brain Barrier Dysfunction. Front Immunol (2021) 12:754141. 10.3389/fimmu.2021.754141
164.
Sladden TM Yerkovich S Grant M Zhang F Liu X Trotter M et al Endothelial Glycocalyx Shedding Predicts Donor Organ Acceptability and Is Associated With Primary Graft Dysfunction in Lung Transplant Recipients. Transplantation (2019) 103:1277–85. 10.1097/TP.0000000000002539
165.
Nelson A Berkestedt I Schmidtchen A Ljunggren L Bodelsson M . Increased Levels of Glycosaminoglycans During Septic Shock: Relation to Mortality and the Antibacterial Actions of Plasma. Shock (2008) 30:623–7. 10.1097/SHK.0b013e3181777da3
166.
Chappell D Jacob M Hofmann-Kiefer K Rehm M Welsch U Conzen P et al Antithrombin Reduces Shedding of the Endothelial Glycocalyx Following Ischaemia/reperfusion. Cardiovasc Res (2009) 83:388–96. 10.1093/cvr/cvp097
167.
Salmon AHJ Neal CR Sage LM Glass CA Harper SJ Bates DO . Angiopoietin-1 Alters Microvascular Permeability Coefficients In Vivo via Modification of Endothelial Glycocalyx. Cardiovasc Res (2009) 83:24–33. 10.1093/cvr/cvp093
168.
Masola V Onisto M Zaza G Lupo A Gambaro G . A New Mechanism of Action of Sulodexide in Diabetic Nephropathy: Inhibits Heparanase-1 and Prevents FGF-2-Induced Renal Epithelial-Mesenchymal Transition. J Transl Med (2012) 10:213. 10.1186/1479-5876-10-213
169.
Siren EMJ Luo HD Tam F Montgomery A Enns W Moon H et al Prevention of Vascular-Allograft Rejection by Protecting the Endothelial Glycocalyx With Immunosuppressive Polymers. Nat Biomed Eng (2021) 5:1202–16. 10.1038/s41551-021-00777-y
170.
Laumonier T Mohacsi PJ Matozan KM Banz Y Haeberli A Korchagina EY et al Endothelial Cell Protection by Dextran Sulfate: A Novel Strategy to Prevent Acute Vascular Rejection in Xenotransplantation. Am J Transpl (2004) 4:181–7. 10.1046/j.1600-6143.2003.00306.x
171.
Laumonier T Walpen AJ Maurus CF Mohacsi PJ Matozan KM Korchagina EY et al Dextran Sulfate Acts as an Endothelial Cell Protectant and Inhibits Human Complement and Natural Killer Cell-Mediated Cytotoxicity against Porcine Cells. Transplantation (2003) 76:838–43. 10.1097/01.TP.0000078898.28399.0A
172.
Banz Y Rieben R Zobrist C Meier P Shaw S Lanz J et al Addition of Dextran Sulfate to Blood Cardioplegia Attenuates Reperfusion Injury in a Porcine Model of Cardiopulmonary Bypass. Eur J Cardiothorac Surg34, 653–60. 10.1016/j.ejcts.2008.05.0242008).
173.
Banz Y Hess OM Robson SC Mettler D Meier P Haeberli A et al Locally Targeted Cytoprotection With Dextran Sulfate Attenuates Experimental Porcine Myocardial Ischaemia/reperfusion Injury. Eur Heart J (2005) 26:2334–43. 10.1093/eurheartj/ehi421
174.
Laumonier T Walpen AJ Matozan KM Korchagina EY Bovin NV Haeberli A et al Multimeric Tyrosine Sulfate Acts as an Endothelial Cell Protectant and Prevents Complement Activation in Xenotransplantation Models. Xenotransplantation (2004) 11:262–8. 10.1111/j.1399-3089.2004.00125.x
Summary
Keywords
xenotransplantation, endothelium, endothelial glycocalyx, complement and coagulation, plasma cascades
Citation
Gultom M and Rieben R (2024) Complement, Coagulation, and Fibrinolysis: The Role of the Endothelium and Its Glycocalyx Layer in Xenotransplantation. Transpl Int 37:13473. doi: 10.3389/ti.2024.13473
Received
01 July 2024
Accepted
27 September 2024
Published
15 October 2024
Volume
37 - 2024
Updates
Copyright
© 2024 Gultom and Rieben.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert Rieben, robert.rieben@unibe.ch
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.